Papillary Renal Cell Carcinomas Rewire Glutathione Metabolism and Are Deficient In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Landscape of Cancer Cell Line Metabolism
The Landscape of Cancer Cell Line Metabolism The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Li, Haoxin, Shaoyang Ning, Mahmoud Ghandi, Gregory V. Kryukov, Shuba Gopal, Amy Deik, Amanda Souza, et. al. 2019. The Landscape of Cancer Cell Line Metabolism. Nature Medicine 25, no. 5: 850-860. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:41899268 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Nat Med Manuscript Author . Author manuscript; Manuscript Author available in PMC 2019 November 08. Published in final edited form as: Nat Med. 2019 May ; 25(5): 850–860. doi:10.1038/s41591-019-0404-8. The Landscape of Cancer Cell Line Metabolism Haoxin Li1,2, Shaoyang Ning3, Mahmoud Ghandi1, Gregory V. Kryukov1, Shuba Gopal1, Amy Deik1, Amanda Souza1, Kerry Pierce1, Paula Keskula1, Desiree Hernandez1, Julie Ann4, Dojna Shkoza4, Verena Apfel5, Yilong Zou1, Francisca Vazquez1, Jordi Barretina4, Raymond A. Pagliarini4, Giorgio G. Galli5, David E. Root1, William C. Hahn1,2, Aviad Tsherniak1, Marios Giannakis1,2, Stuart L. Schreiber1,6, Clary B. Clish1,*, Levi A. Garraway1,2,*, and William R. Sellers1,2,* 1Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA 2Department -

Integrative Pathway Analysis of Metabolic Signature in Bladder Cancer: a Linkage to the Cancer Genome Atlas Project and Prediction of Survival
Integrative Pathway Analysis of Metabolic Signature in Bladder Cancer: A Linkage to The Cancer Genome Atlas Project and Prediction of Survival Friedrich-Carl von Rundstedt,* Kimal Rajapakshe,* Jing Ma, James M. Arnold, Jie Gohlke, Vasanta Putluri, Rashmi Krishnapuram, D. Badrajee Piyarathna, Yair Lotan, Daniel Godde,€ Stephan Roth, Stephan Storkel,€ Jonathan M. Levitt, George Michailidis, Arun Sreekumar, Seth P. Lerner, Cristian Coarfa† and Nagireddy Putluri† From the Scott Department of Urology (FCvR, JML, SPL), Department of Molecular and Cell Biology, Alkek Center for Molecular Discovery (KR, JMA, JG, RK, DBP, AS, CC, NP), Verna and Marrs McLean Department of Biochemistry (AS), Advanced Technology Core (VP, DBP, AS, NP) and Department of Pathology and Immunology (JML), Baylor College of Medicine, Houston and Department of Urology, University of Texas Southwestern Medical Center (YL), Dallas, Texas, Departments of Urology (FCvR, SR) and Pathology Helios Klinikum (DG, SS), Witten-Herdecke University, Wuppertal, Germany, and Department of Statistics, University of Michigan (JM, GM), Ann Arbor, Michigan Purpose: We used targeted mass spectrometry to study the metabolic fingerprint of urothelial cancer and determine whether the biochemical pathway analysis Abbreviations and Acronyms gene signature would have a predictive value in independent cohorts of patients ¼ with bladder cancer. BCa bladder cancer Materials and Methods: Pathologically evaluated, bladder derived tissues, including benign adjacent tissue from 14 patients and bladder cancer from 46, were analyzed by liquid chromatography based targeted mass spectrometry. Differen- tial metabolites associated with tumor samples in comparison to benign tissue were identified by adjusting the p values for multiple testing at a false discovery rate threshold of 15%. -

RT² Profiler PCR Array (Rotor-Gene® Format) Human Amino Acid Metabolism I
RT² Profiler PCR Array (Rotor-Gene® Format) Human Amino Acid Metabolism I Cat. no. 330231 PAHS-129ZR For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Rotor-Gene Q, other Rotor-Gene cyclers Format R Description The Human Amino Acid Metabolism I RT² Profiler PCR Array profiles the expression of 84 key genes important in biosynthesis and degradation of functional amino acids. Of the 20 amino acids required for protein synthesis, six of them (arginine, cysteine, glutamine, leucine, proline, and tryptophan), collectively known as the functional amino acids, regulate key metabolic pathways involved in cellular growth, and development, as well as other important biological processes such as immunity and reproduction. For example, leucine activates mTOR signaling and increases protein synthesis, leading to lymphocyte proliferation. Therefore, a lack of leucine can compromise immune function. Metabolic pathways interrelated with the biosynthesis and degradation of these amino acids include vitamin and cofactor biosynthesis (such as SAM or S-Adenosyl Methionine) as well as neurotransmitter metabolism (such as glutamate). This array includes genes for mammalian functional amino acid metabolism as well as genes involved in methionine metabolism, important also for nutrient sensing and sulfur metabolism. Using realtime PCR, you can easily and reliably analyze the expression of a focused panel of genes involved in functional amino acid metabolism with this array. For further details, consult the RT² Profiler PCR Array Handbook. Shipping and storage RT² Profiler PCR Arrays in the Rotor-Gene format are shipped at ambient temperature, on dry ice, or blue ice packs depending on destination and accompanying products. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

Small-Molecule Binding Sites to Explore New Targets in the Cancer Proteome
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2016 Small-molecule binding sites to explore new targets in the cancer proteome David Xu, Shadia I. Jalal, George W. Sledge Jr., and Samy O. Meroueh* Supplementary Text Druggable Binding Sites across all 10 Diseases. Using the previously established cutoffs, we identified genes that were overexpressed across multiple cancer types and featured druggable binding sites. We ranked these genes based on the total number of tumors that overexpressed the gene (Fig. S1). Using a simple PubMed query, we then counted the number of articles in which either the gene symbol or gene name was co-mentioned with the term ‘cancer’. Most of the most frequently occurring differentially-expressed genes correspond to proteins of well- established cancer targets. Among them are matrix metalloproteinases (MMPs), including MMP1, MMP9, and MMP12, which are implicated in tumor invasion and metastasis (1). There are several protein kinases, including TTK, AURKA, AURKB, and PLK1, that are involved in cell signaling and well-established oncology targets (2). Some genes among this list that have not been extensively studied nor targeted in cancer. These include the serine/threonine kinase PKMYT1 (MYT1) is a regulator of G2/M transition in the cell cycle, but lacks focused small molecule inhibitors that specifically target the kinase. Recent efforts in developing small molecule inhibitors involve repurposing of available kinase inhibitors to specifically target the kinase (3). A subunit of the GINS complex GINS2 (PSF2) is involved in cell proliferation and survival in cancer cell lines (4,5). -
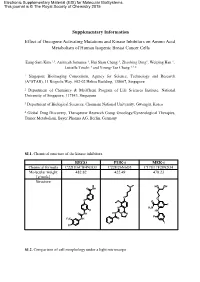
Supplementary Information Effect of Oncogene Activating Mutations And
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2015 Supplementary Information Effect of Oncogene Activating Mutations and Kinase Inhibitors on Amino Acid Metabolism of Human Isogenic Breast Cancer Cells Eung-Sam Kim 1,3, Animesh Samanta 1, Hui Shan Cheng 1, Zhaobing Ding1, Weiping Han 1, Luisella Toschi 4 and Young-Tae Chang 1,2,* 1 Singapore Bioimaging Consortium, Agency for Science, Technology and Research (A*STAR), 11 Biopolis Way, #02-02 Helios Building, 138667, Singapore 2 Department of Chemistry & MedChem Program of Life Sciences Institute, National University of Singapore, 117543, Singapore. 3 Department of Biological Sciences, Chonnam National University, Gwangju, Korea 4 Global Drug Discovery, Therapeutic Research Group Oncology/Gynecological Therapies, Tumor Metabolism, Bayer Pharma AG, Berlin, Germany SI.1. Chemical structure of the kinase inhibitors. REGO PI3K-i MEK-i Chemical formula C22H16ClF4N3O3 C22H26N6O3 C17H17F2IN2O4 Molecular weight 482.82 422.49 478.23 [g/mole] Structure N HO OH O NH O O O O O F N H2N F HN O NH HN N N F F3C NH O N I Cl SI.2. Comparison of cell morphology under a light microscope Scale bar: 100 µm 10x objective lens at Ti microscope (Nikon) SI.3. Scheme of the experimental method for metabolite extraction SI.4. Gradient profile of HPLC run. A : Buffer solution B: Mobile phase H O Time 2 (40 mM NaHPO4, (MeOH/ACN/H2O, Flow rate pH7.8 at RT) 45:45:10, v/v/v) (ml/min) (min) (%) (%) (%) 0 100 0 0 1.0 3 90 10 0 1.0 15 87 13 0 1.0 25 70 30 0 1.0 37 65 35 0 1.0 43 58 42 0 1.0 52 43 57 0 1.0 55 0 100 0 1.0 60 0 0 100 1.0 65 to 80 100 0 0 1.0 SI.5. -

Downloaded from Ftp://Ftp.Uniprot.Org/ on July 3, 2019) Using Maxquant (V1.6.10.43) Search Algorithm
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.17.385096; this version posted November 17, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. The proteomic landscape of resting and activated CD4+ T cells reveal insights into cell differentiation and function Yashwanth Subbannayya1, Markus Haug1, Sneha M. Pinto1, Varshasnata Mohanty2, Hany Zakaria Meås1, Trude Helen Flo1, T.S. Keshava Prasad2 and Richard K. Kandasamy1,* 1Centre of Molecular Inflammation Research (CEMIR), and Department of Clinical and Molecular Medicine (IKOM), Norwegian University of Science and Technology, N-7491 Trondheim, Norway 2Center for Systems Biology and Molecular Medicine, Yenepoya (Deemed to be University), Mangalore, India *Correspondence to: Professor Richard Kumaran Kandasamy Norwegian University of Science and Technology (NTNU) Centre of Molecular Inflammation Research (CEMIR) PO Box 8905 MTFS Trondheim 7491 Norway E-mail: [email protected] (Kandasamy R K) Tel.: +47-7282-4511 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.17.385096; this version posted November 17, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract CD4+ T cells (T helper cells) are cytokine-producing adaptive immune cells that activate or regulate the responses of various immune cells. -

Genomic Evidence of Reactive Oxygen Species Elevation in Papillary Thyroid Carcinoma with Hashimoto Thyroiditis
Endocrine Journal 2015, 62 (10), 857-877 Original Genomic evidence of reactive oxygen species elevation in papillary thyroid carcinoma with Hashimoto thyroiditis Jin Wook Yi1), 2), Ji Yeon Park1), Ji-Youn Sung1), 3), Sang Hyuk Kwak1), 4), Jihan Yu1), 5), Ji Hyun Chang1), 6), Jo-Heon Kim1), 7), Sang Yun Ha1), 8), Eun Kyung Paik1), 9), Woo Seung Lee1), Su-Jin Kim2), Kyu Eun Lee2)* and Ju Han Kim1)* 1) Division of Biomedical Informatics, Seoul National University College of Medicine, Seoul, Korea 2) Department of Surgery, Seoul National University Hospital and College of Medicine, Seoul, Korea 3) Department of Pathology, Kyung Hee University Hospital, Kyung Hee University School of Medicine, Seoul, Korea 4) Kwak Clinic, Okcheon-gun, Chungbuk, Korea 5) Department of Internal Medicine, Uijeongbu St. Mary’s Hospital, Uijeongbu, Korea 6) Department of Radiation Oncology, Seoul St. Mary’s Hospital, Seoul, Korea 7) Department of Pathology, Chonnam National University Hospital, Kwang-Ju, Korea 8) Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea 9) Department of Radiation Oncology, Korea Cancer Center Hospital, Korea Institute of Radiological and Medical Sciences, Seoul, Korea Abstract. Elevated levels of reactive oxygen species (ROS) have been proposed as a risk factor for the development of papillary thyroid carcinoma (PTC) in patients with Hashimoto thyroiditis (HT). However, it has yet to be proven that the total levels of ROS are sufficiently increased to contribute to carcinogenesis. We hypothesized that if the ROS levels were increased in HT, ROS-related genes would also be differently expressed in PTC with HT. To find differentially expressed genes (DEGs) we analyzed data from the Cancer Genomic Atlas, gene expression data from RNA sequencing: 33 from normal thyroid tissue, 232 from PTC without HT, and 60 from PTC with HT. -

Whole-Exome Sequencing Identifies Causative Mutations in Families
BASIC RESEARCH www.jasn.org Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract Amelie T. van der Ven,1 Dervla M. Connaughton,1 Hadas Ityel,1 Nina Mann,1 Makiko Nakayama,1 Jing Chen,1 Asaf Vivante,1 Daw-yang Hwang,1 Julian Schulz,1 Daniela A. Braun,1 Johanna Magdalena Schmidt,1 David Schapiro,1 Ronen Schneider,1 Jillian K. Warejko,1 Ankana Daga,1 Amar J. Majmundar,1 Weizhen Tan,1 Tilman Jobst-Schwan,1 Tobias Hermle,1 Eugen Widmeier,1 Shazia Ashraf,1 Ali Amar,1 Charlotte A. Hoogstraaten,1 Hannah Hugo,1 Thomas M. Kitzler,1 Franziska Kause,1 Caroline M. Kolvenbach,1 Rufeng Dai,1 Leslie Spaneas,1 Kassaundra Amann,1 Deborah R. Stein,1 Michelle A. Baum,1 Michael J.G. Somers,1 Nancy M. Rodig,1 Michael A. Ferguson,1 Avram Z. Traum,1 Ghaleb H. Daouk,1 Radovan Bogdanovic,2 Natasa Stajic,2 Neveen A. Soliman,3,4 Jameela A. Kari,5,6 Sherif El Desoky,5,6 Hanan M. Fathy,7 Danko Milosevic,8 Muna Al-Saffar,1,9 Hazem S. Awad,10 Loai A. Eid,10 Aravind Selvin,11 Prabha Senguttuvan,12 Simone Sanna-Cherchi,13 Heidi L. Rehm,14 Daniel G. MacArthur,14,15 Monkol Lek,14,15 Kristen M. Laricchia,15 Michael W. Wilson,15 Shrikant M. Mane,16 Richard P. Lifton,16,17 Richard S. Lee,18 Stuart B. Bauer,18 Weining Lu,19 Heiko M. Reutter ,20,21 Velibor Tasic,22 Shirlee Shril,1 and Friedhelm Hildebrandt1 Due to the number of contributing authors, the affiliations are listed at the end of this article. -

Metabolic Enzyme Expression Highlights a Key Role for MTHFD2 and the Mitochondrial Folate Pathway in Cancer
ARTICLE Received 1 Nov 2013 | Accepted 17 Dec 2013 | Published 23 Jan 2014 DOI: 10.1038/ncomms4128 Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer Roland Nilsson1,2,*, Mohit Jain3,4,5,6,*,w, Nikhil Madhusudhan3,4,5, Nina Gustafsson Sheppard1,2, Laura Strittmatter3,4,5, Caroline Kampf7, Jenny Huang8, Anna Asplund7 & Vamsi K. Mootha3,4,5 Metabolic remodeling is now widely regarded as a hallmark of cancer, but it is not clear whether individual metabolic strategies are frequently exploited by many tumours. Here we compare messenger RNA profiles of 1,454 metabolic enzymes across 1,981 tumours spanning 19 cancer types to identify enzymes that are consistently differentially expressed. Our meta- analysis recovers established targets of some of the most widely used chemotherapeutics, including dihydrofolate reductase, thymidylate synthase and ribonucleotide reductase, while also spotlighting new enzymes, such as the mitochondrial proline biosynthetic enzyme PYCR1. The highest scoring pathway is mitochondrial one-carbon metabolism and is centred on MTHFD2. MTHFD2 RNA and protein are markedly elevated in many cancers and correlated with poor survival in breast cancer. MTHFD2 is expressed in the developing embryo, but is absent in most healthy adult tissues, even those that are proliferating. Our study highlights the importance of mitochondrial compartmentalization of one-carbon metabolism in cancer and raises important therapeutic hypotheses. 1 Unit of Computational Medicine, Department of Medicine, Karolinska Institutet, 17176 Stockholm, Sweden. 2 Center for Molecular Medicine, Karolinska Institutet, 17176 Stockholm, Sweden. 3 Broad Institute, Cambridge, Massachusetts 02142, USA. 4 Department of Systems Biology, Harvard Medical School, Boston, Massachusetts 02115, USA. -

Glutaminase Is Essential for the Growth of Triple-Negative Breast
RESEARCH ARTICLE Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition Michael Lampa1, Heike Arlt1, Timothy He1, Beatriz Ospina1, Jason Reeves1, Bailin Zhang1, Joshua Murtie1, Gejing Deng1, Claude Barberis2, Dietmar Hoffmann1, Hong Cheng1, a1111111111 Jack Pollard1, Christopher Winter1, Victoria Richon1, Carlos Garcia-Escheverria3, a1111111111 Francisco Adrian1, Dmitri Wiederschain1, Lakshmi Srinivasan1* a1111111111 a1111111111 1 Oncology, Sanofi, Cambridge, MA, United States of America, 2 Integrated Drug Discovery Platform, Sanofi, Waltham, MA, United States of America, 3 Research Platforms, Sanofi, Vitry-Sur-Seine, Vitry, a1111111111 FRANCE * [email protected] OPEN ACCESS Abstract Citation: Lampa M, Arlt H, He T, Ospina B, Reeves J, Zhang B, et al. (2017) Glutaminase is essential Tumor cells display fundamental changes in metabolism and nutrient uptake in order to uti- for the growth of triple-negative breast cancer cells lize additional nutrient sources to meet their enhanced bioenergetic requirements. Gluta- with a deregulated glutamine metabolism pathway mine (Gln) is one such nutrient that is rapidly taken up by tumor cells to fulfill this increased and its suppression synergizes with mTOR inhibition. PLoS ONE 12(9): e0185092. https://doi. metabolic demand. A vital step in the catabolism of glutamine is its conversion to glutamate org/10.1371/journal.pone.0185092 by the mitochondrial enzyme glutaminase (GLS). This study has identified GLS a potential Editor: Ming Tan, University of South Alabama, therapeutic target in breast cancer, specifically in the basal subtype that exhibits a deregu- UNITED STATES lated glutaminolysis pathway. Using inducible shRNA mediated gene knockdown, we dis- Received: June 6, 2017 covered that loss of GLS function in triple-negative breast cancer (TNBC) cell lines with a deregulated glutaminolysis pathway led to profound tumor growth inhibition in vitro and in Accepted: September 6, 2017 vivo.