Density Functional Theory Study of Silica Zeolite Structures: Stabilities and Mechanical Properties of SOD, LTA, CHA, MOR, and MFI
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CO2 Redox Reaction at High-Pressure and High-Temperature D
UCLA UCLA Previously Published Works Title Correspondence: Strongly-driven Re+CO2 redox reaction at high-pressure and high- temperature. Permalink https://escholarship.org/uc/item/93t104f7 Journal Nature communications, 7(1) ISSN 2041-1723 Authors Santamaria-Perez, D McGuire, C Makhluf, A et al. Publication Date 2016-11-29 DOI 10.1038/ncomms13647 Peer reviewed eScholarship.org Powered by the California Digital Library University of California DOI: 10.1038/ncomms13647 OPEN Correspondence: Strongly-driven Re þ CO2 redox reaction at high-pressure and high-temperature D. Santamaria-Perez1,2, C. McGuire1, A. Makhluf1, A. Kavner1, R. Chulia´-Jordan2, J.L. Jorda3,F.Rey3, J. Pellicer-Porres2, D. Martinez-Garcı´a2, P. Rodriguez-Herna´ndez4 & A. Mun˜oz4 Nature Communications 7:13647 doi: 10.1038/ncomms13647 (2016); Published 29 Nov 2016 The discovery of non-molecular carbon dioxide phases under intensities were used to infer the structure. The average formula high pressure and temperature conditions with carbon tetra- of Si0.4C0.6O2 was consistent with X-ray diffraction and Raman 1–3 hedrally coordinated by oxygen atoms has shown that the spectroscopy results. This solid solution based on [CO4] high-density phase diagram of this important substance presents tetrahedra was stable after quenching P-T down to ambient remarkable analogies with those of other group IV oxides. These conditions. results have triggered a variety of experimental studies aiming to We have recently studied the high-pressure chemistry of the explore the high-pressure high-temperature phase diagram of the CO2–SiO2 system up to 50 GPa and 2,400 K using double-sided CO2–SiO2 system, with a goal to find potentially stable laser heating in diamond-anvil cells, while characterizing the compounds. -

Investigation of Zeolite Systems: Focus on Fenton Chemistry Oxidative Stress from Asbestos Like Minerals and Zeolite-Based Dissolved Oxygen Sensing
INVESTIGATION OF ZEOLITE SYSTEMS: FOCUS ON FENTON CHEMISTRY OXIDATIVE STRESS FROM ASBESTOS LIKE MINERALS AND ZEOLITE-BASED DISSOLVED OXYGEN SENSING DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Toni Ann Ruda ***** The Ohio State University 2007 Dissertation Committee: Approved by Prof. Prabir K. Dutta, Advisor Prof. Susan Olesik _________________________________ Prof. W. James Waldman Advisor Graduate Program in Chemistry Prof. Bernard LaLonde ABSTRACT The research in this dissertation has focused on studying zeolite systems in two areas. The first goal of the research was to examine particle properties leading to hydroxyl radical related toxicity. The second goal was to create an efficient optical oxygen sensor for intracellular monitoring of oxygen. Asbestos is known to cause a variety of respiratory health problems; however, the exact mechanism leading to these problems is unknown. Typically, asbestos participates in Fenton chemistry producing hydroxyl radicals which lead to toxicity in vivo. During these times of oxidative stress in vivo, the intracellular oxygen is under flux. Being able to monitor the intracellular oxygen concentration can provide critical information related to the condition of the cell. Initially, studies were performed focusing on Fenton chemistry and oxidative stress from asbestos like minerals. Researchers are striving to elucidate an exact mechanism of asbestos toxicity because it has led and is still leading to multiple respiratory health problems. The physicochemical characteristics of asbestos contributing to respiratory health problems are not fully defined. The goal of this research was to correlate particle toxicity with physicochemical characteristics to help eventually elucidate the mechanism of asbestos toxicity. -

Silicon Carbonate Phase Formed from Carbon Dioxide and Silica Under Pressure
Silicon carbonate phase formed from carbon dioxide and silica under pressure Mario Santoroa,b,1, Federico Gorellia,b, Julien Hainesc, Olivier Cambonc, Claire Levelutd, and Gaston Garbarinoe aEuropean Laboratory for Nonlinear Spectroscopy, Via Nello Carrara 1, 50019 Sesto Fiorentino, Italy; bIstituto per i Processi Chimico-Fisici–Consiglio Nazionale delle Ricerche, Unità Organizzativa di Supporto di Roma, Piazzale Aldo Moro 2,00185 Rome, Italy; cInstitut Charles Gerhardt Montpellier, Equipe Chimie et Cristallochimie des Matériaux, Unité Mixte de Recherche 5253, Centre National de la Recherche Scientifique, Université Montpellier 2, Place Eugène Bataillon, cc1504, 34095 Montpellier Cedex 5, France; dLaboratoire Charles Coulomb, Unité Mixte de Recherche 5221, Centre National de la Recherche Scientifique, Université Montpellier 2, Place Eugène Bataillon, cc026, 34095 Montpellier Cedex 5, France; and eEuropean Synchrotron Radiation Facility, 38343 Grenoble, France Edited by Russell J. Hemley, Carnegie Institution of Washington, Washington, DC, and approved March 31, 2011 (received for review December 30, 2010) The discovery of nonmolecular carbon dioxide under high-pressure density functional theory (DFT) simulations (17). The choice of conditions shows that there are remarkable analogies between silicalite is motivated by the large effective surface exposed to the this important substance and other group IV oxides. A natural and CO2 in the pores (all of the SiO4 tetrahedra are on the surface of long-standing question is whether compounds between CO2 and the micropores), which is likely to be a crucial factor for enhan- SiO2 are possible. Under ambient conditions, CO2 and SiO2 are ther- cing the chemical reaction. modynamically stable and do not react with each other. We show that reactions occur at high pressures indicating that silica can Results and Discussion behave in a manner similar to ionic metal oxides that form carbo- We examined chemical reactions between silicalite SiO2 and nates at room pressure. -

Heterogeneous Catalyst Deactivation and Regeneration: a Review
Catalysts 2015, 5, 145-269; doi:10.3390/catal5010145 OPEN ACCESS catalysts ISSN 2073-4344 www.mdpi.com/journal/catalysts Review Heterogeneous Catalyst Deactivation and Regeneration: A Review Morris D. Argyle and Calvin H. Bartholomew * Chemical Engineering Department, Brigham Young University, Provo, UT 84602, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel: +1-801-422-4162, Fax: +1-801-422-0151. Academic Editor: Keith Hohn Received: 30 December 2013 / Accepted: 12 September 2014 / Published: 26 February 2015 Abstract: Deactivation of heterogeneous catalysts is a ubiquitous problem that causes loss of catalytic rate with time. This review on deactivation and regeneration of heterogeneous catalysts classifies deactivation by type (chemical, thermal, and mechanical) and by mechanism (poisoning, fouling, thermal degradation, vapor formation, vapor-solid and solid-solid reactions, and attrition/crushing). The key features and considerations for each of these deactivation types is reviewed in detail with reference to the latest literature reports in these areas. Two case studies on the deactivation mechanisms of catalysts used for cobalt Fischer-Tropsch and selective catalytic reduction are considered to provide additional depth in the topics of sintering, coking, poisoning, and fouling. Regeneration considerations and options are also briefly discussed for each deactivation mechanism. Keywords: heterogeneous catalysis; deactivation; regeneration 1. Introduction Catalyst deactivation, the loss over time of catalytic activity and/or selectivity, is a problem of great and continuing concern in the practice of industrial catalytic processes. Costs to industry for catalyst replacement and process shutdown total billions of dollars per year. Time scales for catalyst deactivation vary considerably; for example, in the case of cracking catalysts, catalyst mortality may be on the order of seconds, while in ammonia synthesis the iron catalyst may last for 5–10 years. -

A Database of Sources of Information on Mineral Reaction Kinetics
A database of sources of information on mineral reaction kinetics Internal Report IR/05/051 BRITISH GEOLOGICAL SURVEY INTERNAL REPORT IR/05/051 A database of sources of information on mineral reaction kinetics C.A. Rochelle and G. Turner Key words Kinetics, rate, dissolution, precipitation, database, EndNote, literature, searchable. Bibliographical reference ROCHELLE, C.A. AND TURNER G. 2005. A database of sources of information on mineral reaction kinetics. British Geological Survey Internal Report, IR/05/051. 106 pp. © NERC 2005 Keyworth, Nottingham British Geological Survey 2005 BRITISH GEOLOGICAL SURVEY The full range of Survey publications is available from the Keyworth, Nottingham NG12 5GG BGS Sales Desks at Nottingham and Edinburgh; see 0115-936 3241 Fax 0115-936 3488 contact details below or shop online at www.thebgs.co.uk e-mail: [email protected] The London Information Office maintains a reference www.bgs.ac.uk collection of BGS publications including maps for Shop online at: www.thebgs.co.uk consultation. The Survey publishes an annual catalogue of its maps and Murchison House, West Mains Road, Edinburgh EH9 other publications; this catalogue is available from any of 3LA the BGS Sales Desks. The British Geological Survey carries out the geological 0131-667 1000 Fax 0131-668 2683 survey of Great Britain and Northern Ireland (the latter as e-mail: [email protected] an agency service for the government of Northern Ireland), and of the surrounding continental shelf, as well as its London Information Office at the Natural History basic research projects. It also undertakes programmes of Museum (Earth Galleries), Exhibition Road, South British technical aid in geology in developing countries as Kensington, London SW7 2DE arranged by the Department for International Development 020-7589 4090 Fax 020-7584 8270 and other agencies. -

A Partial Glossary of Spanish Geological Terms Exclusive of Most Cognates
U.S. DEPARTMENT OF THE INTERIOR U.S. GEOLOGICAL SURVEY A Partial Glossary of Spanish Geological Terms Exclusive of Most Cognates by Keith R. Long Open-File Report 91-0579 This report is preliminary and has not been reviewed for conformity with U.S. Geological Survey editorial standards or with the North American Stratigraphic Code. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government. 1991 Preface In recent years, almost all countries in Latin America have adopted democratic political systems and liberal economic policies. The resulting favorable investment climate has spurred a new wave of North American investment in Latin American mineral resources and has improved cooperation between geoscience organizations on both continents. The U.S. Geological Survey (USGS) has responded to the new situation through cooperative mineral resource investigations with a number of countries in Latin America. These activities are now being coordinated by the USGS's Center for Inter-American Mineral Resource Investigations (CIMRI), recently established in Tucson, Arizona. In the course of CIMRI's work, we have found a need for a compilation of Spanish geological and mining terminology that goes beyond the few Spanish-English geological dictionaries available. Even geologists who are fluent in Spanish often encounter local terminology oijerga that is unfamiliar. These terms, which have grown out of five centuries of mining tradition in Latin America, and frequently draw on native languages, usually cannot be found in standard dictionaries. There are, of course, many geological terms which can be recognized even by geologists who speak little or no Spanish. -

Mechanism of Chemical Reactions Between Sio2 and CO2 Under Mantle Conditions † † ‡ § § ∥ Xue Yong, John S
Article Cite This: ACS Earth Space Chem. XXXX, XXX, XXX−XXX Mechanism of Chemical Reactions between SiO2 and CO2 under Mantle Conditions † † ‡ § § ∥ Xue Yong, John S. Tse,*, , , and Jiuhua Chen , † Department of Physics and Engineering Physics, University of Saskatchewan, Saskatoon, Saskatchewan, Canada S7N 5E2 ‡ State Key Laboratory for Superhard Materials, Jilin University, Changchun, China 130012 § Center for High Pressure Science and Technology Advanced Research, Changchun, China 130015 ∥ Center for the Study of Matter at Extreme Conditions, Florida International University, Miami, Florida 33199, United States *S Supporting Information ABSTRACT: Silica (SiO2) is a major component of many minerals on the Earth. Under ambient conditions, silica does not react with carbon dioxide (CO2). However, at high pressure and temperature, the stability ff fl of silica may be a ected by CO2, which becomes supercritical uid CO2 under extreme conditions and can percolate into the Earth’s mantle to react with silica. Here, we investigated the chemical reactions between zeolite SSZ-56 as a model silicate and CO2 under temperature and pressure conditions close to those orf the mantle transition zone using density functional theory and molecular dynamics calculations. The ’ reactions occurred on the SiO2 surface forming the zeolite s cavities. In the melt, CO2 and SiO2 mixed closely and, upon cooling, formed a solid with disordered Si and C sites similar to a cristobalite − α SiO2 CO2 solid-solution structure. This structure was thermodynamically stable with respect to -cristobalite and solid CO2 above 9 GPa. KEYWORDS: silica, DFT, chemical reaction, high pressure, Earth’s mantle, mineral 4−6 1. INTRODUCTION CO2 from the atmosphere. -

United States Patent (19) (11) 4,061,724 Grose Et Al
United States Patent (19) (11) 4,061,724 Grose et al. - 45) Dec. 6, 1977 (54. CRYSTALLINE SILICA OTHER PUBLICATIONS (75) Inventors: Robert William Grose, Mahopac; Merrill et al., "J. Phys. Chem...,' 55, 187, (1951). Edith Marie Flanigen, White Plains, both of N.Y. Primary Examiner-Jack Cooper Attorney, Agent, or Firm-Richard G. Miller 73) Assignee: Union Carbide Corporation, New 57 ABSTRACT York, N.Y. A novel crystalline silica composition having uniform (21) Appl. No.: 615,557 pore dimensions of approximately 6 Angstrom units is prepared by calcining a crystalline hydrated alkylonium 22 Filed: Sept. 22, 1975 silicate prepared hydrothermally from a reaction mix ture containing as essential reagents, water, amorphous 51) Int. Cl? .............................................. C01B33/12 silica and a quaternary ammonium compound at a pH of 52 U.S. C. ..................................... 423/335; 423/339 at least 10. The crystalline silica, because of its uniform 58) Field of Search ................................ 423/335,339 pore structure, is capable of making size-selective sepa rations of molecular species, but neither it nor its silicate (56) References Cited precursor exhibition-exchange properties. U.S. PATENT DOCUMENTs This absorbent has a very useful hydrophobic/organo 3,556,725 1/1971 Chiola et al.......................... 423/339 philic characteristic which permits its use in selectively adsorbing organic materials from water, either in liquid FOREIGN PATENT DOCUMENTS or vapor phase. 1,003,195 2/1957 Germany ............................. 423/339 927,658 5/1963 United Kingdom ................. 423/339 2 Claims, No Drawings 4,061,724 1. 2 TABLE I-continued CRYSTALLINE SLCA DENSITY AND REFRACTIVE INPR: DATA - FOR CRYSTALLINE SILICAS - - The present invention relates in general to a novel Silica D d, g/cc crystalline silica composition and to the method for 5 logite Keatite 1.513, 1.522 2.50 preparing same. -

Application of Mineral Sorbents for Removal of Petroleum Substances: a Review
minerals Review Application of Mineral Sorbents for Removal of Petroleum Substances: A Review Lidia Bandura 1,*, Agnieszka Woszuk 2, Dorota Kołody ´nska 3 and Wojciech Franus 1 1 Department of Geotechnical Science, Faculty of Civil Engineering and Architecture, Lublin University of Technology, Nadbystrzycka 40, 20-618 Lublin, Poland; [email protected] 2 Department of Roads and Bridges, Faculty of Civil Engineering and Architecture, Lublin University of Technology, Nadbystrzycka 40, 20-618 Lublin, Poland; [email protected] 3 Department of Inorganic Chemistry, Faculty of Chemistry, Maria Curie-Skłodowska University, Maria Curie-Skłodowska Sq.2, 20-031 Lublin, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-509-381-660 Academic Editor: Annalisa Martucci Received: 15 December 2016; Accepted: 4 March 2017; Published: 8 March 2017 Abstract: Environmental pollution with petroleum products has become a major problem worldwide, and is a consequence of industrial growth. The development of sustainable methods for the removal of petroleum substances and their derivatives from aquatic and terrestrial environments and from air has therefore become extremely important today. Advanced technologies and materials dedicated to this purpose are relatively expensive; sorption methods involving mineral sorbents are therefore popular and are widely described in the scientific literature. Mineral materials are easily available, low-cost, universal adsorbents and have a number of properties that make them suitable for the removal of petroleum substances. This review describes recent works on the use of natural, synthetic and modified mineral adsorbents for the removal of petroleum substances and their derivatives from roads, water and air. Keywords: petroleum substances; oils; BTEX; sorption; removal; mineral adsorbents 1. -
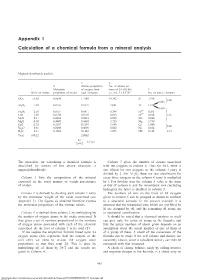
Appendix 1 Calculation of a Chemical Formula from a Mineral Analysis
Appendix 1 Calculation of a chemical formula from a mineral analysis Appendix 1 Magnesiohornblende analysis 3 4 2 Atomic proportion No. of anions on 1 Molecular of oxygen from basis of 24 (O,OH) 5 Wt.% of oxides proportion of oxides each molecule i.e. col. 368.3735 No. of ions in formula SiO 51.63 0.8594 1.7188 14.392 Si 7.196 2 8.00 0.804 } Al2O3 7.39 0.0725 0.2175 1.821 Al 1.214 0.410 3+ Fe2O3 2.50 0.0157 0.0471 0.394 Fe 0.263 FeO 5.30 0.0738 0.0738 0.618 Fe2+ 0.618 5.07 MnO 0.17 0.0024 0.0024 0.020 Mn 0.020 } MgO 18.09 0.4489 0.4489 3.759 Mg 3.759 CaO 12.32 0.2197 0.2197 1.840 Ca 1.840 2.00 Na2O 0.61 0.0098 0.0098 0.082 Na 0.164 } H2O+ 2.31 0.1282 0.1282 1.073 OH 2.146 2.15 Total 100.32 2.8662 24 = 8.3735 2.8662 The procedure for calculating a chemical formula is Column 5 gives the number of cations associated described by means of the above example, a with the oxygens in column 4. Thus for SiO2 there is magnesiohornblende. one silicon for two oxygens so the column 4 entry is divided by 2. For A12O3 there are two aluminiums for Column 1 lists the composition of the mineral every three oxygens so the column 4 entry is multiplied expressed in the usual manner as weight percentages by ~˜. -

A Reevaluation of the Calorimetric Data for the Enthalpy of Formation of Some K- and Na-Bearing Silicate Minerals1
UNITED STATES DEPARTMENT OF THE INTERIOR U.S. GEOLOGICAL SURVEY A reevaluation of the calorimetric data for the enthalpy of formation of some K- and Na-bearing silicate minerals1 by B. S. Hemingway2 and H. T. Haselton, Jr. Open-file Report 94-576 1 This report is preliminary and has not been edited or reviewed for conformity with U.S. Geological Survey editorial standards. 2 U. S. Geological Survey, 959 National Center, Reston, VA 22092 Abstract Calorimetric data from aqueous HF and molten-salt calorimetry have been reviewed for the phases microcline, sanidine, muscovite, analcime, sodium aluminate, low albite, jadeite, and analbite. The review was conducted to evaluate the suggestion by Sverjensky et al. (1991) that the calorimetric data for K- and Na-bearing phases was in error by -6.69 and -6.80 kJ'mol"1 , respectively; and the suggestion by Johnson et al. (1992) that silicalite replace quartz as the Si reference phase in aqueous HF calorimetry. Our examination of the data set does not support either suggestion. Our recommended values for the enthalpies of formation from the elements at 298.15 K for muscovite, analcime, sodium aluminate, low albite, jadeite, and analbite (-5974.8 ± 4.9, -3309.8 ±3.3, -1135.6 ± 1.4, -3935.0 ± 2.6, -3029.3 ± 2.5, and -3923.6 ± 2.6 kJ'mol" 1 ) are essentially identical to the generally accepted calorimetric values given by Hemingway and Robie (1977) or Robie et al. (1979). However, our recommended values for the enthalpy of formation from the elements of microcline and sanidine are -7.2 and -6.3 kJ more stable than the values given by those sources. -

Computational Studies of Silica
Computational studies of silica Kathryn Glaser Department of Chemistry, University College London May 2011 Supervisors: Dr. R. G. Bell, Prof. C. R. A. Catlow and Prof. A. Takada A thesis submitted for the degree of Doctor of Engineering Declaration I, Kathryn Joan Glaser confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 1 Abstract There are three areas of research in this thesis. The first is con- cerned with the silica polymorph, tridymite, with simulations car- ried out using three computational methods: free energy minimisa- tion, molecular dynamics and Density Functional Theory. A num- ber of tridymite structures with different atomic configurations have been found in nature. The simulations explore various properties of these different forms of tridymite and investigate whether it is pos- sible to distinguish between them using the three computational techniques. It was found that the interatomic potential and simula- tion technique used, rather than the simulation temperature, were the main factors affecting the resulting structure. There are a num- ber of possible explanations for this result: The techniques may not be sensitive enough to deal with an energy landscape as flat as in the case of tridymite. Another reason is that the potentials have been parameterised to distinguish between structures which have recon- structive transitions (where bonds are broken and formed) and may not be able to deal with displacive transitions (where only angles between atoms change) as with tridymite. The final possible expla- nation is that a number of the known structures may be meta-stable and/or poorly characterised.