Identification of Genetic Heterogeneity in a South Indian Family Affected By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human Artificial Chromosome (Hac) Vector
Europäisches Patentamt *EP001559782A1* (19) European Patent Office Office européen des brevets (11) EP 1 559 782 A1 (12) EUROPEAN PATENT APPLICATION published in accordance with Art. 158(3) EPC (43) Date of publication: (51) Int Cl.7: C12N 15/09, C12N 1/15, 03.08.2005 Bulletin 2005/31 C12N 1/19, C12N 1/21, C12N 5/10, C12P 21/02 (21) Application number: 03751334.8 (86) International application number: (22) Date of filing: 03.10.2003 PCT/JP2003/012734 (87) International publication number: WO 2004/031385 (15.04.2004 Gazette 2004/16) (84) Designated Contracting States: • KATOH, Motonobu, Tottori University AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Yonago-shi, Tottori 683-8503 (JP) HU IE IT LI LU MC NL PT RO SE SI SK TR • TOMIZUKA, Kazuma, Designated Extension States: Kirin Beer Kabushiki Kaisha AL LT LV MK Takashi-shi, Gunma 370-1295 (JP) • KUROIWA, Yoshimi, (30) Priority: 04.10.2002 JP 2002292853 Kirin Beer Kabushiki Kaisha Takasaki-shi, Gunma 370-1295 (JP) (71) Applicant: KIRIN BEER KABUSHIKI KAISHA • KAKEDA, Minoru, Kirin Beer Kabushiki Kaisha Tokyo 104-8288 (JP) Takasaki-shi, Gunma 370-1295 (JP) (72) Inventors: (74) Representative: HOFFMANN - EITLE • OSHIMURA, Mitsuo, Tottori University Patent- und Rechtsanwälte Yonago-shi, Tottori 683-8503 (JP) Arabellastrasse 4 81925 München (DE) (54) HUMAN ARTIFICIAL CHROMOSOME (HAC) VECTOR (57) The present invention relates to a human arti- ing a cell which expresses foreign DNA. Furthermore, ficial chromosome (HAC) vector and a method for pro- the present invention relates to a method for producing ducing the same. -

(12) United States Patent (10) Patent No.: US 8,703,482 B2 Oshimura Et Al
USOO8703482B2 (12) United States Patent (10) Patent No.: US 8,703,482 B2 Oshimura et al. (45) Date of Patent: Apr. 22, 2014 (54) HUMAN ARTIFICIAL CHROMOSOME (HAC) Rasheed et al. “Characterization of a Newly Derived Human Sar VECTOR coma Cell Line (HT-1080).” Cancer, 1974, vol. 33, pp. 1027-1033. R. Moreadith etal, “Gene targeting in embryonic stem cells: the new physiology and metabolism.” J Mol Med, 1997, vol. 75, pp. 208-216. (75) Inventors: Mitsuo Oshimura, Tottori (JP); L. Mullins et al., “Perspective Series: Molecular Medicine in Geneti Motonobu Katoh, Tottori (JP); Kazuma cally Engineered Animals: Transgenesis in the Rat and Larger Ani Tomizuka, Gunma (JP); Yoshimi mals,” Journal of Clinical Investigation, 1996, vol. 97, No. 7, pp. Kuroiwa, Gunma (JP); Minoru 1557-1560. Kakeda, Gunma (JP) M. Pera et al. “Human embryonic stem cells,” Journal of Cell Sci ence, 2000, vol. 113, pp. 5-10. Hattori et al. “The DNA sequence of human chromosome 21.” (73) Assignee: Kyowa Hakko Kirin Co., Ltd., Tokyo Nature, 2000, vol. 405, No. 6784, pp. 311-319. (JP) European Search Report dated May 18, 2006 for counterpart appli cation PCT/JP0312734 to parent U.S. Appl. No. 10/530.207. (*) Notice: Subject to any disclaimer, the term of this Y. Kuroiwa et al. “Efficient modification of a human chromosome by patent is extended or adjusted under 35 telomere-directed truncation in high homologous recombination U.S.C. 154(b) by 176 days. proficient chicken DT40 cells.” Nucleic Acids. Res., 1998, vol. 26, No. 14, pp. 3447-3448. Y. Kuroiwa et al., “Manipulation of human minichromosomes to carry (21) Appl. -

A Study of the Differential Expression Profiles of Keshan Disease Lncrna/Mrna Genes Based on RNA-Seq
421 Original Article A study of the differential expression profiles of Keshan disease lncRNA/mRNA genes based on RNA-seq Guangyong Huang1, Jingwen Liu2, Yuehai Wang1, Youzhang Xiang3 1Department of Cardiology, Liaocheng People’s Hospital of Shandong University, Liaocheng, China; 2School of Nursing, Liaocheng Vocational & Technical College, Liaocheng, China; 3Shandong Institute for Endemic Disease Control, Jinan, China Contributions: (I) Conception and design: G Huang, Y Xiang; (II) Administrative support: G Huang, Y Wang; (III) Provision of study materials or patients: G Huang, J Liu, Y Xiang; (IV) Collection and assembly of data: G Huang, J Liu; (V) Data analysis and interpretation: J Liu, Y Wang; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Guangyong Huang, MD, PhD. Department of Cardiology, Liaocheng People’s Hospital of Shandong University, No. 67 of Dongchang Street, Liaocheng 252000, China. Email: [email protected]. Background: This study aims to analyze the differential expression profiles of lncRNA in Keshan disease (KSD) and to explore the molecular mechanism of the disease occurrence and development. Methods: RNA-seq technology was used to construct the lncRNA/mRNA expression library of a KSD group (n=10) and a control group (n=10), and then Cuffdiff software was used to obtain the gene lncRNA/ mRNA FPKM value as the expression profile of lncRNA/mRNA. The fold changes between the two sets of samples were calculated to obtain differential lncRNA/mRNA expression profiles, and a bioinformatics analysis of differentially expressed genes was performed. Results: A total of 89,905 lncRNAs and 20,315 mRNAs were detected. -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Lupus Nephritis Supp Table 5
Supplementary Table 5 : Transcripts and DAVID pathways correlating with the expression of CD4 in lupus kidney biopsies Positive correlation Negative correlation Transcripts Pathways Transcripts Pathways Identifier Gene Symbol Correlation coefficient with CD4 Annotation Cluster 1 Enrichment Score: 26.47 Count P_Value Benjamini Identifier Gene Symbol Correlation coefficient with CD4 Annotation Cluster 1 Enrichment Score: 3.16 Count P_Value Benjamini ILMN_1727284 CD4 1 GOTERM_BP_FAT translational elongation 74 2.50E-42 1.00E-38 ILMN_1681389 C2H2 zinc finger protein-0.40001984 INTERPRO Ubiquitin-conjugating enzyme/RWD-like 17 2.00E-05 4.20E-02 ILMN_1772218 HLA-DPA1 0.934229063 SP_PIR_KEYWORDS ribosome 60 2.00E-41 4.60E-39 ILMN_1768954 RIBC1 -0.400186083 SMART UBCc 14 1.00E-04 3.50E-02 ILMN_1778977 TYROBP 0.933302249 KEGG_PATHWAY Ribosome 65 3.80E-35 6.60E-33 ILMN_1699190 SORCS1 -0.400223681 SP_PIR_KEYWORDS ubl conjugation pathway 81 1.30E-04 2.30E-02 ILMN_1689655 HLA-DRA 0.915891173 SP_PIR_KEYWORDS protein biosynthesis 91 4.10E-34 7.20E-32 ILMN_3249088 LOC93432 -0.400285215 GOTERM_MF_FAT small conjugating protein ligase activity 35 1.40E-04 4.40E-02 ILMN_3228688 HLA-DRB1 0.906190291 SP_PIR_KEYWORDS ribonucleoprotein 114 4.80E-34 6.70E-32 ILMN_1680436 CSH2 -0.400299744 SP_PIR_KEYWORDS ligase 54 1.50E-04 2.00E-02 ILMN_2157441 HLA-DRA 0.902996561 GOTERM_CC_FAT cytosolic ribosome 59 3.20E-33 2.30E-30 ILMN_1722755 KRTAP6-2 -0.400334007 GOTERM_MF_FAT acid-amino acid ligase activity 40 1.60E-04 4.00E-02 ILMN_2066066 HLA-DRB6 0.901531942 SP_PIR_KEYWORDS -

Diversity and Regulatory Impact of Copy Number Variation in the Primate Macaca Fascicularis
Gschwind et al. BMC Genomics (2017) 18:144 DOI 10.1186/s12864-017-3531-y RESEARCHARTICLE Open Access Diversity and regulatory impact of copy number variation in the primate Macaca fascicularis Andreas R. Gschwind1,2, Anjali Singh3, Ulrich Certa3, Alexandre Reymond1* and Tobias Heckel3* Abstract Background: Copy number variations (CNVs) are a significant source of genetic diversity and commonly found in mammalian genomes. We have generated a genome-wide CNV map for Cynomolgus monkeys (Macaca fascicularis). This crab-eating macaque is the closest animal model to humans that is used in biomedical research. Results: We show that Cynomolgus monkey CNVs are in general much smaller in size than gene loci and are specific to the population of origin. Genome-wide expression data from five vitally important organs demonstrates that CNVs in close proximity to transcription start sites associate strongly with expression changes. Among these eQTL genes we find an overrepresentation of genes involved in metabolism, receptor activity, and transcription. Conclusion: These results provide evidence that CNVs shape tissue transcriptomes in monkey populations, potentially offering an adaptive advantage. We suggest that this genetic diversity should be taken into account when using Cynomolgus macaques as models. Keywords: Cynomolgus monkey, CNV, Gene expression, eQTL, Olfactory receptors Background genes they can alter gene dosage, disrupt coding se- Copy number variations (CNVs) are genetic differences quences or modify the level and timing of gene expres- in the normal population displayed as microscopically sion for genes within the CNV [9, 10] and on its flanks invisible deletions or amplifications of stretches of gen- [4, 11–15]. -

Supplementary Data
SUPPLEMENTARY METHODS 1) Characterisation of OCCC cell line gene expression profiles using Prediction Analysis for Microarrays (PAM) The ovarian cancer dataset from Hendrix et al (25) was used to predict the phenotypes of the cell lines used in this study. Hendrix et al (25) analysed a series of 103 ovarian samples using the Affymetrix U133A array platform (GEO: GSE6008). This dataset comprises clear cell (n=8), endometrioid (n=37), mucinous (n=13) and serous epithelial (n=41) primary ovarian carcinomas and samples from 4 normal ovaries. To build the predictor, the Prediction Analysis of Microarrays (PAM) package in R environment was employed (http://rss.acs.unt.edu/Rdoc/library/pamr/html/00Index.html). When more than one probe described the expression of a given gene, we used the probe with the highest median absolute deviation across the samples. The dataset from Hendrix et al. (25) and the dataset of OCCC cell lines described in this manuscript were then overlaid on the basis of 11536 common unique HGNC gene symbols. Only the 99 primary ovarian cancers samples and the four normal ovary samples were used to build the predictor. Following leave one out cross-validation, a predictor based upon 126 genes was able to identify correctly the four distinct phenotypes of primary ovarian tumour samples with a misclassification rate of 18.3%. This predictor was subsequently applied to the expression data from the 12 OCCC cell lines to determine the likeliest phenotype of the OCCC cell lines compared to primary ovarian cancers. Posterior probabilities were estimated for each cell line in comparison to the following phenotypes: clear cell, endometrioid, mucinous and serous epithelial. -

An Evolutionary Medicine Perspective on Neandertal Extinction
Supplementary Information (Figures and Tables) for: An evolutionary medicine perspective on Neandertal extinction Alexis P. Sullivan1, Marc de Manuel3, Tomas Marques-Bonet3,4,5, & George H. Perry1,2 Departments of 1Biology and 2Anthropology, Pennsylvania State University, University Park, PA 16802, USA 3Institut de Biologia Evolutiva (CSIC/UPF), Parque de Investigación Biomédica de Barcelona (PRBB), Barcelona, Catalonia 08003, Spain 4CNAG-CRG, Centre for Genomic Regulation (CRG), Barcelona Institute of Science and Technology (BIST), Baldiri i Reixac 4, 08028 Barcelona, Spain 5Catalan Institution of Research and Advanced Studies (ICREA), Passeig de Lluís Companys, 23, 08010, Barcelona, Spain Corresponding Author: George H. Perry E-mail: [email protected] Supplemental Figure 1: Innate immune system gene permutation analyses – 10,000 sets of 73 randomly selected genes containing nonsynonymous SNPs Supplemental Figure 2: Virus-interacting protein gene permutation analyses – 10,000 sets of 164 randomly selected genes containing nonsynonymous SNPs Supplemental Figure 3: MHC gene permutation analyses – 10,000 sets of 13 randomly selected genes containing nonsynonymous SNPs Supplemental Figure 4: Patterns of Neandertal and modern human nonsynonymous SNP diversity in MHC genes (n = 13) excluding the Altai Neandertal and one random modern human per population Supplemental Figure 5: Significantly enriched gene ontology categories (red) among top 1% ape diversity genes Supplemental Table 1: A comparison of genome-wide nonsynonymous SNPs versus total (nonsynonymous -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e . -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -

Epigenetic Silencing Mediated Through Activated PI3K/AKT Signaling in Breast Cancer
SUPPLEMENTARY: Epigenetic Silencing Mediated Through Activated PI3K/AKT Signaling in Breast Cancer Tao Zuo, Ta-Ming Liu, Xun Lan, Yu-I Weng, Rulong Shen, Fei Gu, Yi-Wen Huang, Sandya Liyanarachchi, Daniel E. Deatherage, Pei-Yin Hsu, Cenny Taslim, Bhuvaneswari Ramaswamy, Charles L. Shapiro, Huey-Jen L. Lin, Alfred S. L. Cheng, Victor X. Jin, and Tim H.-M. Huang 1 Table of Content: 1. Fig. S1. Elevated AKT1 kinase activity in myrAKT1- transfected MCF10A cells. ---------------------------------------------------------------------------------------------------- page 4 2. Fig. S2. Genome-wide view of H3K27me3 in mammary epithelial cells. ---------------------------------------------------------------------------------------------------- page 5 3. Fig. S3. Biological functions of PI3K/AKT- downstream genes. ----------------- page 6 4. Fig. S4. Genomic maps of 40 genes. ------------------------------------------------- page 8 5. Fig. S5. Comparing DNA methylation levels of four CpG islands measured by meDIP-qPCR and MassARRAY.------------------------------------------------------- page 11 6. Fig. S6. Validation of DNA methylation by MBDCap-seq.----------------------- page 13 7. Table S1. Summary of H3K27me3 ChIP-seq data ------------------------------ page 14 8. Table S2. Distribution of H3K27me3 in MCF10A genome ---------------------- page 14 9. Table S3. Genes (n=488) enriched with H3K27me3 and downregulated by PI3K/AKT- signaling ---------------------------------------------------------------------- page 15 10. Table S4. Genes (n=1005) enriched -
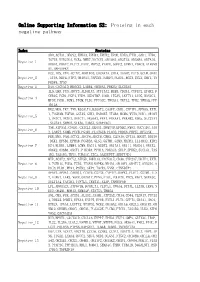
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2