View Annual Report
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Annual Report
Fiscal Year 2015 Annual Report To: Governor Charlie Baker Secretary of Administration and Finance Kristen Lepore Secretary of Housing & Economic Development Jay Ash Senate President Stan Rosenberg Speaker of the House Robert DeLeo State Comptroller Thomas G. Shack III Clerk of the Senate William Welch Clerk of the House of Representatives Steven James By forward: House and Senate Committees on Ways and Means and the Joint Committee on Economic Development and Emerging Technologies Date: September 30, 2015 Re: FY 2015 Annual Report of the Massachusetts Life Sciences Center The Massachusetts Life Sciences Center (MLSC) respectfully submits this Annual Report detailing the organization’s operations and accomplishments during Fiscal Year (FY) 2015. For the past seven years, the MLSC has led the state’s life sciences initiative – a $1 billion initiative designed to strengthen the state’s economy by accelerating the growth of our life sciences sectors in Massachusetts. The MLSC’s goal has been to make Massachusetts the strongest life sciences ecosystem in the world and the best place in the world for life sciences companies to do business. The MLSC collaborates with the private sector to accelerate the pace of innovation and economic development in all regions of Massachusetts. The MLSC’s strategy has been based on the belief that the best role for the state is to invest in Massachusetts’ innovation capacity – the ability to produce and commercialize a flow of innovative life sciences technologies over the long term. Since 2008, the MLSC has invested aggressively in five “enablers” that build innovation capacity: translational research, entrepreneurship, workforce development, infrastructure and collaboration. -

Annual Report
HUMAN CAPITAL FISCAL YEAR 2019 ANNUAL REPORT INNOVATION CAPITAL GROWTH CAPITAL INTELLECTUAL CAPITAL The capital of scientific revolution. Fiscal Year 2019 Annual Report • Massachusetts Life Sciences Center • i BOARD OF DIRECTORS TABLE OF CONTENTS A LETTER FROM THE INTERIM PRESIDENT & CEO Michael J. Heffernan Co-Chair; Secretary, Executive Office for Administration & Finance A Letter from the President & CEO ...........................................................................................1 Mike Kennealy Co-Chair; Secretary, Executive Office Vision & Strategy: The Capital of Scientific Revolution ......................................................2 The Patients Are Waiting of Housing & Economic Development Fiscal Year 2019 Highlights and The Bottom Line ..............................................................3 Gary P. Kearney, MD Around the globe, billions of patients and their loved ones await advances in health President, Longwood Urological Associates Human Capital care that will produce the products, devices, and therapies to alleviate suffering, Marty Meehan Internship Challenge: Enhancing Career Exploration improve treatment, and save lives. Much of that attention falls on Massachusetts, as President, University of Massachusetts and Expanding the Talent Pipeline ............................................................................................4 the world looks to our ecosystem to deliver the breakthroughs that further unlock our Peter Parker understanding of human physiology, harness the power of data science, -

Science-USA (Boston+), November 2012
Schweizerische Eidgenossenschaft Confédération suisse Confederazione Svizzera Confederaziun svizra Science-USA (Boston+), November 2012 Table of Contents 1. Policy ........................................................................................................................................................................................ 1 2. Education .................................................................................................................................................................................. 2 3. Life Science .............................................................................................................................................................................. 3 4. Nano / Micro Technology / Material Science ............................................................................................................................. 4 5. Information & Communications Technology ............................................................................................................................. 6 6. Energy / Environment ............................................................................................................................................................... 6 7. Engineering / Robotics / Space ................................................................................................................................................. 8 8. Physics / Chemistry / Math ...................................................................................................................................................... -

LIVIO VALENTI CV 2011-Present VAXESS TECHNOLOGIES, INC
LIVIO VALENTI CV Livio Valenti is the co-founder of Vaxess Technologies and Giorgio Ruffolo Research Fellow in the Sustainability Science Program at Harvard Kennedy School of Government and the Harvard Global Health Institute. He received a Master in Public Policy from Harvard’s Kennedy School of Government in 2013, where he was the recipient of the Empedocle Maffia Fellowship provided by the Italian Government to one outstanding individual to attend Harvard. While a student at Harvard, he co-founded Vaxess Technologies, an innovative life sciences company that aims to create vaccines that do not need refrigeration. Vaxess has raised $4.75m in venture capital, angel investors, and various public institutions, including the Massachusetts Life Science Center (MLSC). Harvard Business School (HBS) published a case on Vaxess in 2014. Livio was named by Forbes one of the 30 under 30 individuals likely to change the world for his commitment in improving global health and one of the 10 under 40 (PrimiDieci-Under40 2014) most distinguished individuals with Italian origins or Italian nationality currently in the USA by the Italian-American Chamber of Commerce and the Italian Ministry of Foreign Affairs. His academic research at Harvard focuses on the space at the intersection between Government and Business. Livio is researching the potential to sustainably commercialize innovative technologies to improve global health. He is contributing to collaborative work with the Initiative on Innovation and Access to Technologies for Sustainable Development led by William Clark. Professional experience Prior to studying at Harvard, Livio worked for the United Nations in several developing countries. His work included advising government clients and international financing institutions in the identification, appraisal and financing of large investment projects. -

Genocea Biosciences, Inc. (Name of Registrant As Specified in Its Charter)
UNITED STATES SECURITIES AND EXCHANGE COMMISSION Washington, D.C. 20549 SCHEDULE 14A Proxy Statement Pursuant to Section 14(a) of the Securities Exchange Act of 1934 Filed by the Registrant ☒ Filed by a Party other than the Registrant o Check the appropriate box: o Preliminary Proxy Statement o Confidential, for Use of the Commission Only (as permitted by Rule 14a-6(e)(2)) ☒ Definitive Proxy Statement o Definitive Additional Materials o Soliciting Material under §240.14a-12 Genocea Biosciences, Inc. (Name of Registrant as Specified In Its Charter) (Name of Person(s) Filing Proxy Statement, if other than the Registrant) Payment of Filing Fee (Check the appropriate box): ☒ No fee required. o Fee computed on table below per Exchange Act Rules 14a-6(i)(1) and 0-11. (1) Title of each class of securities to which transaction applies: (2) Aggregate number of securities to which transaction applies: Per unit price or other underlying value of transaction computed pursuant to Exchange Act Rule 0-11 (set forth the amount on which the (3) filing fee is calculated and state how it was determined): (4) Proposed maximum aggregate value of transaction: (5) Total fee paid: o Fee paid previously with preliminary materials. Check box if any part of the fee is offset as provided by Exchange Act Rule 0-11(a)(2) and identify the filing for which the offsetting fee o was paid previously. Identify the previous filing by registration statement number, or the Form or Schedule and the date of its filing. (1) Amount Previously Paid: (2) Form, Schedule or Registration Statement No.: (3) Filing Party: (4) Date Filed: 1 Genocea Biosciences, Inc. -

RENT a BENCH Biotech Incubators Such As Labcentral Are Lowering Barriers to Entrepreneurship by Trisha Gura, in Cambridge, Massachusetts
NEWS | FEATURES | ENTREPRENEURSHIP GOT A STARTUP? RENT A BENCH Biotech incubators such as LabCentral are lowering barriers to entrepreneurship By Trisha Gura, in Cambridge, Massachusetts ith its cookie-cutter windows and square-meter facility, now owned by the boxy brick exterior, the build- Massachusetts Institute of Technology ing here on Main Street could be (MIT) and leased to LabCentral, functions just another former 19th century as a life science “incubator” that helps bud- W warehouse or factory. It’s no sur- ding biotech firms combat the soaring costs prise to learn that passenger rail- of lab space and equipment in the red-hot road cars were once built in its spacious Boston-Cambridge region. Any scientist on June 11, 2015 interior. Yet the Cambridge landmark has with an idea and ambition can rent a bench a history not just of production, but also and an office, sharing space, services, and of innovation. In 1876, it housed the office high-cost tools with others pursuing their of Thomas Watson, assistant to Alexander own entrepreneurial dreams. “It is very ex- Graham Bell, when the pair made the first citing because we are there at the nascent two-way long-distance phone call, to Bos- moment of many really, really cool com- ton. In the 20th century, it was home to Po- panies,” says molecular biologist Johannes laroid and the lab of Edwin Land when he Fruehauf, a LabCentral founder. invented the instant camera. It’s also the nascent moment for facili- www.sciencemag.org Today, a nonprofit organization called ties like LabCentral. -
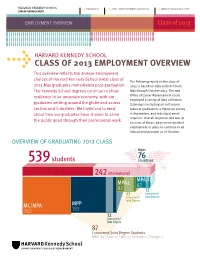
Class of 2013 Employment Overview 76
HARVARD KENNEDY SCHOOL 617-495-1161 [email protected] www.hks.harvard.edu/career CAREER ADVANCEMENT t t t EMPLOYMENT OVERVIEW Class of 2013 HARVARD KENNEDY SCHOOL t CLASS OF 2013 EMPLOYMENT OVERVIEW This overview reflects the diverse employment choices of Harvard Kennedy School (hks) class of The following report on the class of 2013 May graduates immediately post-graduation. 2013 is based on data collected from The Kennedy School degrees continue to show May through October 2013. The hks resilience in an uncertain economy, with our Office of Career Advancement (oca) employed a variety of data collection graduates settling around the globe and across techniques including an exit survey sectors and industries. We invite you to read taken at graduation, a follow-up survey about how our graduates have chosen to serve in September, and individual email inquiries. Overall response rate was 91 the public good through their professional work. percent; of those, 90 percent reported employment or plans to continue in an educational program as of October. OVERVIEW OF GRADUATING 2013 CLASS from 76 539 students countries 242 international MPA/ID MPA2 62 82 12 43 Concurrent/ Concurrent/ Joint Degree Joint Degree MC/MPA MPP 202 193 32 Concurrent/ Joint Degree 87 Concurrent/Joint Degree Students MBA: 69 / Law: 11 / MD: 5 / Divinity: 1 / Design: 1 HARVARD KENNEDY SCHOOL 617-495-1161 [email protected] www.hks.harvard.edu/career CAREER ADVANCEMENT t t t EMPLOYMENT OVERVIEW Class of 2013 HIGHLIGHTS GRADUATES t Eight graduates were selected as t Nonprofit/ngo employment is down IN 2013 finalists in the U.S. -

Fiscal Year 2015 Annual Report
Fiscal Year 2015 Annual Report To: Governor Charlie Baker Secretary of Administration and Finance Kristen Lepore Secretary of Housing & Economic Development Jay Ash Senate President Stan Rosenberg Speaker of the House Robert DeLeo State Comptroller Thomas G. Shack III Clerk of the Senate William Welch Clerk of the House of Representatives Steven James By forward: House and Senate Committees on Ways and Means and the Joint Committee on Economic Development and Emerging Technologies Date: September 30, 2015 Re: FY 2015 Annual Report of the Massachusetts Life Sciences Center The Massachusetts Life Sciences Center (MLSC) respectfully submits this Annual Report detailing the organization’s operations and accomplishments during Fiscal Year (FY) 2015. For the past seven years, the MLSC has led the state’s life sciences initiative – a $1 billion initiative designed to strengthen the state’s economy by accelerating the growth of our life sciences sectors in Massachusetts. The MLSC’s goal has been to make Massachusetts the strongest life sciences ecosystem in the world and the best place in the world for life sciences companies to do business. The MLSC collaborates with the private sector to accelerate the pace of innovation and economic development in all regions of Massachusetts. The MLSC’s strategy has been based on the belief that the best role for the state is to invest in Massachusetts’ innovation capacity – the ability to produce and commercialize a flow of innovative life sciences technologies over the long term. Since 2008, the MLSC has invested aggressively in five “enablers” that build innovation capacity: translational research, entrepreneurship, workforce development, infrastructure and collaboration. -

What's Inside
Newsletter A publication of the Controlled Release Society Volume 34 • Number 2 • 2017 What’s Inside Formulation and Delivery of Mono-N-Oxide Vinblastine Liposomal Formulation for Targeting Non-Small Cell Lung Cancer with Hypoxic Environments In Vitro and In Vivo Evaluation of Controlled Payload Release Using Acoustically Responsive Scaffolds CRS Annual Meeting DDTR Update Science. People. Products. Connect and collaborate with delivery science and technology leaders from academia and industry, under one roof at #CRSboston Join us in Boston, Massachusetts, U.S.A. from July 16–19 for the 2017 Controlled Release Society Annual Meeting & Exposition. Registration is now open! Visit controlledreleasesociety.org/CRSboston for details. 2 Newsletter Ryan Donnelly Vol. 34 • No. 2 • 2017 Editor > TABLE OF CONTENTS 4 From the Editor 5 2017 Controlled Release Society Annual Meeting & Exposition Steven Giannos 7 Scientifically Speaking Editor Formulation and Delivery of Mono-N-Oxide Vinblastine Liposomal Formulation for Targeting Non-Small Cell Lung Cancer with Hypoxic Environments 10 Scientifically Speaking In Vitro and In Vivo Evaluation of Controlled Payload Release Using Acoustically Responsive Scaffolds 13 DDTR Update Drug Delivery and Translational Research Update Medha Joshi Editor 14 People in the News 15 Companies in the News Cover image: Malignant effusion: Pleural fluid cytology of lung (pulmonary) papillary adenocarcinoma, a type of non small cell carcinoma. Pap stain. David Litman / Shutterstock.com Arlene McDowell Editor Bozena Michniak-Kohn Editor Rod Walker Editor 3 > FROM THE EDITOR Editors Ryan Donnelly Steven Giannos Steven Giannos Medha Joshi University of Texas Medical Branch Arlene McDowell Galveston, TX, U.S.A. Bozena Michniak-Kohn Rod Walker The CRS Newsletter is the official Please Come to Boston newsletter of the Controlled Release Society. -

Resi Program Final High
redefining eARLY sTAGE iNVESTMENTs RESI 3 Early Stage Life Science Investment Just Hit a Home Run! ONSITE GUIDE September 17, 2014 Fenway Park Boston, MA RESIConference.com #RESI3 LifeScienceNation.com EMC Level FENWAY PARK EMC CLUB 3 LEVEL 3 EMC2 SUITE L2 Exit FIELD VIEW Board Room Exit Exit Stairs/Escalators workshopS TO State st. Bar Area pavilion LEVEL PARTNERING Forum REGISTRATION sTATE sTREET PAVILION Level State Street. PAVILION lEVEL 4 FENWAY PARK pRESS ROOM mEDIA dINING TRACK 2 TRACK 1 Exit 13 1 2 12 1 FIELD VIEW 2 12 3 Exit Exit 11 11 3 4 10 10 4 5 5 6 6 7 7 8 9 9 8 15 25 16 25 16 26 32 17 30 17 26 27 28 29 30 31 18 24 27 28 29 18 Stairs/Escalators 24 19 23 23 22 22 21 21 20 20 19 13 14 14 15 Bar Area PANELS Exhibiting companies TO EMC LEVEL pRESENTING companies contents TABLE OF CONTENTS Welcome from Dennis Ford - - - - - - - 2 RESI Schedule - - - - - - 3 Investor Panels - - - - - - - 5 Early Stage Workshops - - - - - - 25 Partnering Forum - - - - - - - 26 RESI Innovation Challenge - - - - - - - 27 Exhibiting Companies - - - - - - 29 Sponsors - - - - - - - 30 Media Partners - - - - - - - 33 1 welcome to resi Life Science Nation would like to welcome you to the third Redening Early Stage Investments (RESI) conference. We're excited to see you here at our new landmark location, Fenway Park. I hope you are ready to get the most out of a day lled with content, meetings and opportunities for everyone who has a stake in the early-stage life science sector. -

Vaxess' Long Road to Heat-Stable Vaccines
Case Number 2081.0 Technological Innovation for Global Health: Vaxess’ Long Road to Heat-Stable Vaccines It was only April, but 2015 was shaping up to be a busy one for Livio Valenti. The Harvard Kennedy School graduate was named to Forbes’ prestigious “30 under 30” list of the brightest entrepreneurs in science and healthcare, just as Vaxess Technologies, the biotech start-up he co-founded in 2012, was starting to attract atten- tion. Indeed, the company was awarded a research grant from the U.S. National Science Foundation and the U.S. National Institute of Health to put toward the development of heat-stable vaccines—a major honor and accom- plishment, though the grants themselves were modest. Most vaccines had to be refrigerated (or frozen) to preserve their efficacy. However, keeping vaccines at the appropriate temperature from manufacturer to recipient posed significant costs and logistical challenges, especial- ly in many low- and middle-income countries where the majority of the world’s children lived. Valenti believed Vaxess’ technology could provide a potentially simpler, safer approach to heat-stabilizing vaccines from both a scientific and regulatory standpoint—formulating them to withstand a lack of refrigeration—compared to other preservation techniques. In the vaccine field however, the risks and costs of research and development, and lengthy regulatory approval processes were major challenges to advancing products to market. Furthermore, with the greatest need for heat-stable vaccines in developing countries, market incentives for companies to invest in heat-stabilization research and development (R&D) were weak. Still, the U.S. Food and Drug Administration (FDA) had approved silk fibroin—the key ingredient for Vaxess’ platform to heat-stabilize vaccines—for use in medical devices such as surgical sutures and implantable surgical mesh, giving the Vaxess team confidence that their inno- vation would eventually gain FDA approval. -

Vaxess Technologies Closes Final Tranche of $8.2 Million Series a Funding
Vaxess Technologies closes final tranche of $8.2 million Series A funding 01 July 2019 | News | By Prapti Shah It appoints Dr. Purnanand Sarma to Board Vaxess Technologies, a biotechnology company redefining drug delivery with the silk-powered MIMIX smart release patch has announced it has closed the final tranche of an $8.2 million Series A round. Led by The Engine, the venture capital firm launched by MIT in 2016 to invest in early-stage Tough Tech companies, the round includes participation from Bioinnovation Capital. To date, Vaxess has raised more than $22 million in both grant and equity funding, including backing from the Bill and Melinda Gates Foundation, the National Science Foundation and the National Institutes of Health. Additionally, Vaxess has announced the appointment of drug delivery and development veteran Dr. Purnanand Sarma to the board of directors. Dr. Sarma brings extensive experience in all aspects of the pharmaceutical business. He currently serves as the President and Chief Executive Officer of Immunome, a biotechnology company developing first-in-class therapeutics leveraging the most highly educated components of the immune system from patients. Until recently, he served as the President & CEO of TARIS Biomedical, where he raised more than $100 million in equity financing, drove multiple products into the clinic, and entered into a deal worth up to $587 million with Allergan all based on the company’s proprietary sustained release drug delivery system. Vaxess was born out of technologies developed at MIT and Tufts University where researchers discovered that silk fibroin — an essential structural protein in silk — was an optimal building block for advanced biomaterials.