Carbon Dioxide Reduction to Methane and Coupling with Acetylene to Form Propylene Catalyzed by Remodeled Nitrogenase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Conversion of Carbon Dioxide to Acetylene on a Micro Scale
810 NATURE June 14, 1947 Vol. 159 orbitale of the ethmoid is reduced". In the orang Stainless steel was found to be the most satis and the gibbon a large planum orbitale articulates factory furnace material tried. From mild steel in front with the lacrimal, as in man. The figure relatively large amounts of acetylene were produced we give of the orbital wall in Pleaianthropus shows in blank experiments, and a fused silica envelope a condition almost exactly as in man. fitted with a nickel thimble was found, after it had We are here not at present concerned with the been used with calcium and barium metals, to absorb question of whether man and the Australopithecinre carbon dioxide when hot even when no calcium or have arisen from an early anthropoid, or a pre barium was present. In carrying out the absorption anthropoid, or an Old World monkey or a tarsioid ; of carbon dioxide by barium metal in the stainless but we think the evidence afforded by this new skull steel furnace it was found that when the pressure of Plesianthropus shows that the Australopithecinre at which the gas was admitted was less than about and man are very closely allied, and that these small 10·1 mm. of mercury, the yield of acetylene was brained man-like beings were very nearly human. variable and only about 45 per cent. Good yields R. BROOM were obtained when the carbon dioxide at its full J. T. RoBINSON pressure was admitted to the furnace before raising Transvaal Museum, Pretoria. the temperature above 400° C. -
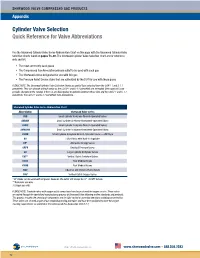
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

Kinetic Study of the Selective Hydrogenation of Acetylene Over Supported Palladium Under Tail-End Conditions
catalysts Article Kinetic Study of the Selective Hydrogenation of Acetylene over Supported Palladium under Tail-End Conditions Caroline Urmès 1,2, Jean-Marc Schweitzer 2, Amandine Cabiac 2 and Yves Schuurman 1,* 1 IRCELYON CNRS, UMR 5256, Univ Lyon, Université Claude Bernard Lyon 1, 2 avenue Albert Einstein, 69626 Villeurbanne Cedex, France; [email protected] 2 IFP Energies nouvelles, Etablissement de Lyon, Rond-point de l’échangeur de Solaize, BP3, 69360 Solaize, France; [email protected] (J.-M.S.); [email protected] (A.C.) * Correspondence: [email protected]; Tel.: +33-472445482 Received: 9 January 2019; Accepted: 31 January 2019; Published: 14 February 2019 Abstract: The kinetics of the selective hydrogenation of acetylene in the presence of an excess of ethylene has been studied over a 0.05 wt. % Pd/α-Al2O3 catalyst. The experimental reaction conditions were chosen to operate under intrinsic kinetic conditions, free from heat and mass transfer limitations. The data could be described adequately by a Langmuir–Hinshelwood rate-equation based on a series of sequential hydrogen additions according to the Horiuti–Polanyi mechanism. The mechanism involves a single active site on which both the conversion of acetylene and ethylene take place. Keywords: power-law; Langmuir–Hinshelwood; kinetic modeling; Pd/α-Al2O3 1. Introduction Ethylene is the largest of the basic chemical building blocks with a global market estimated at more than 140 million tons per year with an increasing growth rate. It is used mainly as precursor for polymers production, for instance polyethylene, vinyl chloride, ethylbenzene, or even ethylene oxide synthesis. -

Common Name: METHYL ACETYLENE HAZARD SUMMARY
Common Name: METHYL ACETYLENE CAS Number: 74-99-7 RTK Substance number: 1218 DOT Number: UN 1954 Date: May 1999 ----------------------------------------------------------------------- ----------------------------------------------------------------------- HAZARD SUMMARY * Methyl Acetylene can affect you when breathed in. * Exposure to hazardous substances should be routinely * Breathing Methyl Acetylene can irritate the nose and evaluated. This may include collecting personal and area throat. air samples. You can obtain copies of sampling results * Breathing Methyl Acetylene can irritate the lungs causing from your employer. You have a legal right to this coughing and/or shortness of breath. Higher exposures information under OSHA 1910.1020. can cause a build-up of fluid in the lungs (pulmonary * If you think you are experiencing any work-related health edema), a medical emergency, with severe shortness of problems, see a doctor trained to recognize occupational breath. diseases. Take this Fact Sheet with you. * Exposure to Methyl Acetylene can cause headache, dizziness, convulsions (fits) and unconsciousness. WORKPLACE EXPOSURE LIMITS * Liquified Methyl Acetylene can cause frostbite. OSHA: The legal airborne permissible exposure limit * Methyl Acetylene is a HIGHLY FLAMMABLE and (PEL) is 1,000 ppm averaged over an 8-hour REACTIVE chemical and is a DANGEROUS FIRE and workshift. EXPLOSION HAZARD. NIOSH: The recommended airborne exposure limit is IDENTIFICATION 1,000 ppm averaged over a 10-hour workshift. Methyl Acetylene is a colorless gas, which is shipped as a liquid, with a sweet odor. It is used as a simple anesthetic and ACGIH: The recommended airborne exposure limit is in the manufacture of other chemicals, and as a welding torch 1,000 ppm averaged over an 8-hour workshift. -

Benzene⋯Acetylene: a Structural Investigation of the Prototypical CH
Physical Chemistry Chemical Physics Benzene ⋯⋯⋯Acetylene: A structural investigation of the prototypical CH ⋯⋯⋯π interaction Journal: Physical Chemistry Chemical Physics Manuscript ID: CP-ART-02-2014-000845.R1 Article Type: Paper Date Submitted by the Author: 20-Mar-2014 Complete List of Authors: Ulrich, Nathan; Eastern Illinois University, Chemistry Seifert, Nathan; University of Virginia, Chemistry Dorris, Rachel; Eastern Illinois University, Chemistry Peebles, Rebecca; Eastern Illinois University, Chemistry Pate, Brookes; University of Virginia, Chemistry Peebles, Sean; Eastern Illinois University, Chemistry Page 1 of 30 Physical Chemistry Chemical Physics 3/20/2014 10:13:21 AM Benzene ⋯⋯⋯A⋯AAAcetylene: A structural investigation of the prototypical CH ⋯⋯⋯π interaction Nathan W. Ulrich, a Nathan A. Seifert, b Rachel E. Dorris, a Rebecca A. Peebles, a Brooks H. Pate, b Sean A. Peebles a, * a Eastern Illinois University, Department of Chemistry, 600 Lincoln Ave., Charleston, IL 61920 b University of Virginia, Department of Chemistry and Biochemistry, McCormick Rd., PO Box 400319, Charlottesville, VA 22904 *To whom correspondence should be addressed: email: [email protected] phone: (217) 581-2679 1 Physical Chemistry Chemical Physics Page 2 of 30 3/20/2014 10:13:21 AM Abstract The structure of a prototype CH ⋯π system, benzene ⋯acetylene, has been determined in the gas phase using Fourier-transform microwave spectroscopy. The spectrum is consistent with an effective C6v structure with an H ⋯π distance of 2.4921(1) Å. The HCCH subunit likely tilts by ~5º from the benzene symmetry axis. The dipole moment was determined to be 0.438(11) D from Stark effect measurements. The observed intermolecular distance is longer than in similar benzene ⋯HX complexes and than the distances observed in the benzene ⋯HCCH cocrystal and predicted by many high level ab initio calculations; however, the experimentally estimated –1 binding energy of 7.1(7) kJ mol is similar to previously studied benzene ⋯HX complexes. -

Acetylene Fact Sheet
Occupational Safety Fact Sheet Compressed Acetylene Gas DESCRIPTION ACETYLENE STORAGE Acetylene is a compressed gas that is commonly Acetylene cylinders must always be transported, used in conjunction with compressed oxygen to fuel stored and used in the upright position. torches used for various tasks. Acetylene cylinders must be stored with the valve HAZARDS caps installed and in a well-ventilated area. Particular care must be taken with small cylinders that do not have valve protection caps. Keep cylinders away from external sources of heat. Cylinders are not designed for temperatures in excess of 125F (52C). Acetylene must be stored at least 20-feet away from oxygen or separated by a five foot high fire-rated wall. MSHA Photo Acetylene poses unique hazards based on its high flammability, instability and unique storage and transportation requirements. Acetylene is highly unstable. High pressure or temperatures can result in decomposition that can result in fire or explosion. Acetylene cylinders must never be transported or stored in a closed vehicle. There are several documented explosions that have occurred due to the buildup of acetylene vapors inside of a vehicle that were triggered by a spark from the vehicle’s Canadian Center for Occupation Health & Safety electrical system. ACETYLENE USE THE UNIQUE ACETYLENE CYLINDER Regulator Connection - Always carefully inspect Acetylene cylinders do not the CGA connection on the cylinder and remove any contain compressed or visible contamination before connecting the liquefied acetylene; instead regulator. they contain acetylene gas dissolved in acetone that is Leak Test - Once all components are connected, absorbed onto a porous mass perform a leak test with a soap and water solution. -

(Pahs) Coming from Pyrolysis in Low-Pressure Gas Carburizing Conditions Tsilla Bensabath, Hubert Monnier, Pierre-Alexandre Glaude
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archive Ouverte en Sciences de l'Information et de la Communication Detailed kinetic modeling of the formation of toxic polycyclic aromatic hydrocarbons (PAHs) coming from pyrolysis in low-pressure gas carburizing conditions Tsilla Bensabath, Hubert Monnier, Pierre-Alexandre Glaude To cite this version: Tsilla Bensabath, Hubert Monnier, Pierre-Alexandre Glaude. Detailed kinetic modeling of the for- mation of toxic polycyclic aromatic hydrocarbons (PAHs) coming from pyrolysis in low-pressure gas carburizing conditions. Journal of Analytical and Applied Pyrolysis, Elsevier, 2016, 122, pp.342-354. 10.1016/j.jaap.2016.09.007. hal-01510237 HAL Id: hal-01510237 https://hal.archives-ouvertes.fr/hal-01510237 Submitted on 19 Apr 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Title: Detailed kinetic modeling of the formation of toxic polycyclic aromatic hydrocarbons (PAHs) coming from pyrolysis in low-pressure gas carburizing conditions Authors: Tsilla Bensabatha,b -

On Reversible H2 Loss Upon N2 Binding to Femo-Cofactor of Nitrogenase
On reversible H2 loss upon N2 binding to FeMo-cofactor of nitrogenase Zhi-Yong Yanga, Nimesh Khadkaa, Dmitriy Lukoyanovb, Brian M. Hoffmanb,1, Dennis R. Deanc,1, and Lance C. Seefeldta,1 aDepartment of Chemistry and Biochemistry, Utah State University, Logan, UT 84322; bDepartments of Chemistry and Molecular Biosciences, Northwestern University, Evanston, IL 60208; and cDepartment of Biochemistry, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061 Contributed by Brian M. Hoffman, August 23, 2013 (sent for review July 15, 2013) Nitrogenase is activated for N2 reduction by the accumulation of obligatory, reversible H2 loss upon N2 binding (5). We consid- four electrons/protons on its active site FeMo-cofactor, yielding ered two models for this process, both built on our character- a state, designated as E4, which contains two iron-bridging hydrides ization of the key E4 state, and tested them against the numerous – – [Fe H Fe]. A central puzzle of nitrogenase function is an apparently constraints imposed by turnover under N2 plus D2 or T2 (5, 9– obligatory formation of one H2 per N2 reduced, which would “waste” 16). In particular, these constraints include the key findings that two reducing equivalents and four ATP. We recently presented during catalytic reduction of N2 (see Scheme S1), a molecule of a draft mechanism for nitrogenase that provides an explanation D2 or T2 will reduce two protons to form two HD or HT without + + for obligatory H2 production. In this model, H2 is produced by re- D /T exchange with solvent, even though neither D2 nor T2 by ductive elimination of the two bridging hydrides of E4 during N2 themselves reacts with nitrogenase during turnover under Ar (5). -

Highly Selective Room Temperature Acetylene Sorption by an Unusual Triacetylenic Phosphine MOF
ChemComm Highly selective room temperature acetylene sorption by an unusual triacetylenic phosphine MOF Journal: ChemComm Manuscript ID CC-COM-07-2018-005402.R1 Article Type: Communication Page 1 of 5 Please doChemComm not adjust margins Journal Name COMMUNICATION Highly selective room temperature acetylene sorption by an unu- sual triacetylenic phosphine MOF† a a ‡,b a ,b Received 00th January 20xx, Joseph E. Reynolds III, Kelly M. Walsh, Bin Li, Pranaw Kunal, Banglin Chen* and Simon M. Accepted 00th January 20xx Humphrey*,a DOI: 10.1039/x0xx00000x www.rsc.org/ The new ligand tris(p-carboxyphenylethynyl)phosphine decoration of MOF pore surfaces with particular organic func- (P{C≡CC6H5-4-CO2H}3) was used to synthesize a permanently porous tional groups can encourage stronger physisorption of adsorb- Mn(II)-based acetylenic phosphine coordination material, PCM-48. ates with similar chemical characteristics, via enhancement of This triply-interpenetrated MOF contains 1-D microchannels that host-guest dipolar and/or van der Waals interactions.6 For ex- are decorated with electron-rich and adsorbate-accessible ample, since C2H2 contains polar C–H groups and an electron- acetylenic moieties and phosphine lone pairs. PCM-48 has a rich C≡C core, it has been shown that MOFs decorated with 3 7 moderate room-temperature C2H2 adsorption capacity (25.54 cm abundant polarizing moieties (e.g., hydrogen bond acceptors) ─1 8 g ) and displays high separation selectivities for C2H2 over CH4 or acetylenic groups enable enhanced C2H2 sorption capacities 7–9 (C2H2/CH4 = 23.3), CO2 (C2H2/CO2 = 4.3), and N2 (C2H2/N2 = 76.9) at and selectivities. -

Measurement of Carbonyl Fluoride, Hydrogen Fluoride, and Other Combustion Byproducts During Fire Suppression Testing by Fourier Transform Infrared Spectroscopy
MEASUREMENT OF CARBONYL FLUORIDE, HYDROGEN FLUORIDE, AND OTHER COMBUSTION BYPRODUCTS DURING FIRE SUPPRESSION TESTING BY FOURIER TRANSFORM INFRARED SPECTROSCOPY Craig S. Miser, and W. Randolph Davis Aberdeen Test Center, Aberdeen Proving Ground, MD and Kevin L. McNesby, Army Research Laboratory, Steven H. Hoke U. S. Army Center for Health Promotion and Preventative Medicine and Michael K. Leonnig, Sci-Tech, Services Inc. ABSTRACT The Fourier transform infrared (FTIR)spectrometer is used for gathering toxic byproduct data in real fire situations using Halon replacement candidate systems. A unique approach is used to obtain carbonyl fluoride calibration data. Hydrogen fluoride can be measured in all systems; carbonyl fluoride can be measured in most systems. Several other methodologies are used simultaneously to measure hydrogen fluoride, and these data are compared. Further work will be done to refine this methodology and in-situ measurement will be employed in the future. INTRODUCTION The United States Army is committed to replacing its halon-based fire suppression systems. Evaluation of candidate replacement systems is a complex problem that requires the analytical measurement of chemical byproducts. Factors such as agent dispersion, extinguishment time, and bottle configuration can all affect the nature and quantity of the gaseous byproducts. Hydrofluorocarbon (HFC) based replacement systems, such as hexafluoropropane, heptafluoropropane, and octafluoropropane, stoichiometrically contain about twice the fluorine of Halon 1301 systems. These HFC-based systems also have extinguishing concentrations roughly twice that of Halon 1301, according to conventional cup burner data [l]. These factors. combine to produce systems with about four times more total fluorine. This causes great concern for the production of carbonyl fluoride (CFzO) and hydrogen fluoride (HF)as toxic byproducts [2]. -

Guidelines for Incompatible Chemicals Chemicals Avoid
Fire Department 401 Oak Street, #402 Roseville, California 95678-2649 Guidelines for Incompatible Chemicals Following is a list of common chemicals followed by substances from which they should be kept separated: Chemicals Avoid Chromic acid, nitric acid, hydroxyl containing Acetic Acid compounds, ethylene glycol, perchloric acid, peroxides and permanganates. Acetone Concentrated nitric and sulfuric mixtures Chlorine, bromine, copper, silver, fluorine and Acetylene mercury Carbon dioxide, carbon tetrachloride and other Alkali and Alkaijne Earth Metals, such as chlorinated hydrocarbons. (Also prohibit water, Sodium Potassium, Cesium, Lithium, foam and dry chemical on fires involving these Magnesium, Calcium also Aluminum metals. Mercury, chlorine, calcium hypochlorite, Iodine, Ammonia (Anhydrous) bromine and hydrogen fluoride, any mineral acids. Acids, metal powders, flammable liquids, Ammonium Nitrate chlorates, nitrates, sulfur, finely divided organics or combustibles Aniline Nitric acid, hydrogen peroxide Antimony Pentasulfide Chlorates, nitrates, other oxidizing agents, acids (Golden Antimony Sulfide) Ammonia, acetylene, butadiene, butane and other petroleum gases, hydrogen, sodium Bromine carbide, turpentine, benzene and finely divided metals Bronze Dust Aluminum Calcium Carbide Water (See also Acetylene) Our Mission… Protect and enhance the safety and well being of residents, businesses, customers and partners. We will accomplish this by…Delivering exceptional service and compassionate solutions as a cohesive team with dedication, -

Polycyclic Aromatic Hydrocarbon (PAH) Formation During Acetylene
Polycyclic aromatic hydrocarbon (PAH) formation during acetylene pyrolysis in tubular reactor under low pressure carburizing conditions Tsilla Bensabath, Minh Duy Le, Hubert Monnier, Pierre-Alexandre Glaude To cite this version: Tsilla Bensabath, Minh Duy Le, Hubert Monnier, Pierre-Alexandre Glaude. Polycyclic aro- matic hydrocarbon (PAH) formation during acetylene pyrolysis in tubular reactor under low pressure carburizing conditions. Chemical Engineering Science, Elsevier, 2019, 202, pp.84-94. 10.1016/j.ces.2019.03.030. hal-02139154 HAL Id: hal-02139154 https://hal.archives-ouvertes.fr/hal-02139154 Submitted on 24 May 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Polycyclic Aromatic Hydrocarbon (PAH) Formation during Acetylene Pyrolysis in Tubular Reactor under Low Pressure Carburizing Conditions Tsilla Bensabath1,2, Minh Duy Le2, Hubert Monnier2,* Pierre-Alexandre Glaude1 1Laboratoire Réactions et Génie des Procédés, CNRS, Université de Lorraine, 1, rue Grandville, 54001 Nancy, France 2Institut National de Recherche et de Sécurité, 1, rue du Morvan, 54519 Vandœuvre-lès- Nancy, France *Corresponding author: Hubert Monnier INRS, Institut National de Recherche et de Sécurité, 1, rue du Morvan, 54519 Vandœuvre-lès- Nancy, France [email protected] Highlights Experiments of acetylene pyrolysis at 1173 K and 8 kPa in a tubular reactor.