Surface States
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Surface Vs Bulk Electronic Structures of a Moderately Correlated
Surface vs bulk electronic structures of a moderately correlated topological insulator YbB6 revealed by ARPES N. Xu,1* C. E. Matt,1,2 E. Pomjakushina,3 J. H. Dil,4,1 G. Landolt, 1,5 J.-Z. Ma,1,6 X. Shi,1,6 R. S. Dhaka,1,4,7 N. C. Plumb,1 M. Radović,1,8 V. N. Strocov, 1 T. K. Kim,9 M. Hoesch, 9 K. Conder,3 J. Mesot,1,2,4 H. Ding,6, 10 and M. Shi1,† 1Swiss Light Source, Paul Scherrer Institut, CH-5232 Villigen PSI, Switzerland 2Laboratory for Solid State Physics, ETH Zürich, CH-8093 Zürich, Switzerland 3Laboratory for Developments and Methods, Paul Scherrer Institut, CH-5232 Villigen PSI, Switzerland 4Institute of Condensed Matter Physics, Ecole Polytechnique Fedé ralé de Lausanne, CH-1015 Lausanne, Switzerland 5Physik-Institut, Universität Zürich, Winterthurerstrauss 190, CH-8057 Zürich, Switzerland 6Beijing National Laboratory for Condensed Matter Physics and Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China 7Department of Physics, Indian Institute of Technology Delhi, Hauz Khas, New Delhi-110016, India 8 SwissFEL, Paul Scherrer Institut, CH-5232 Villigen PSI, Switzerland 9Diamond Light Source, Harwell Science and Innovation Campus, Didcot OX11 0DE, UK, 10 Collaborative Innovation Center of Quantum Matter, Beijing, China Page: 1 Systematic angle-resolved photoemission spectroscopy (ARPES) experiments have been carried out to investigate the bulk and (100) surface electronic structures of a topological mixed-valence insulator candidate, YbB6. The bulk states of YbB6 were probed with bulk-sensitive soft X-ray ARPES, which show strong three-dimensionality as required by cubic symmetry. -

Surface and Quantum-Well States in Ultra Thin Pt Films on the Au(111) Surface
materials Article Formation of Surface and Quantum-Well States in Ultra Thin Pt Films on the Au(111) Surface Igor V. Silkin 1,*, Yury M. Koroteev 1,2,3, Pedro M. Echenique 4,5 and Evgueni V. Chulkov 3,4,5 1 Department of Physics, Tomsk State University, 634050 Tomsk, Russia; [email protected] 2 Institute of Strength Physics and Materials Science, Siberian Branch, Russian Academy of Sciences, 634050 Tomsk, Russia 3 Department of Physics, Saint Petersburg State University, 198504 Saint Petersburg, Russia 4 Donostia International Physics Center (DIPC), 20018 San Sebastian/Donostia, Basque Country, Spain; [email protected] (P.M.E.); [email protected] (E.V.C.) 5 Department of Materials Physics, Materials Physics Center CFM-MPC and Mixed Center CSIC-UPV, 20080 San Sebastian/Donostia, Basque Country, Spain * Correspondence: igor [email protected]; Tel.: +34-943-01-8284 Received: 21 November 2017; Accepted: 7 December 2017; Published: 9 December 2017 Abstract: The electronic structure of the Pt/Au(111) heterostructures with a number of Pt monolayers n ranging from one to three is studied in the density-functional-theory framework. The calculations demonstrate that the deposition of the Pt atomic thin films on gold substrate results in strong modifications of the electronic structure at the surface. In particular, the Au(111) s-p-type Shockley surface state becomes completely unoccupied at deposition of any number of Pt monolayers. The Pt adlayer generates numerous quantum-well states in various energy gaps of Au(111) with strong spatial confinement at the surface. As a result, strong enhancement in the local density of state at the surface Pt atomic layer in comparison with clean Pt surface is obtained. -

Theory and Modeling of Field Electron Emission from Low-Dimensional
Theory and Modeling of Field Electron Emission from Low-Dimensional Electron Systems by Alex Andrew Patterson B.S., Electrical Engineering University of Pittsburgh, 2011 M.S., Electrical Engineering Massachusetts Institute of Technology, 2013 Submitted to the Department of Electrical Engineering and Computer Science in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Electrical Engineering at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY February 2018 c Massachusetts Institute of Technology 2018. All rights reserved. Author.............................................................. Department of Electrical Engineering and Computer Science January 31, 2018 Certified by. Akintunde I. Akinwande Professor of Electrical Engineering and Computer Science Thesis Supervisor Accepted by . Leslie A. Kolodziejski Professor of Electrical Engineering and Computer Science Chair, Department Committee on Graduate Theses 2 Theory and Modeling of Field Electron Emission from Low-Dimensional Electron Systems by Alex Andrew Patterson Submitted to the Department of Electrical Engineering and Computer Science on January 31, 2018, in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Electrical Engineering Abstract While experimentalists have succeeded in fabricating nanoscale field electron emit- ters in a variety of geometries and materials for use as electron sources in vacuum nanoelectronic devices, theory and modeling of field electron emission have not kept pace. Treatments of field emission which address individual deviations of real emitter properties from conventional Fowler-Nordheim (FN) theory, such as emission from semiconductors, highly-curved surfaces, or low-dimensional systems, have been de- veloped, but none have sought to treat these properties coherently within a single framework. As a result, the work in this thesis develops a multidimensional, semi- classical framework for field emission, from which models for field emitters of any dimensionality, geometry, and material can be derived. -

Surface Electronic Structure
Home Search Collections Journals About Contact us My IOPscience Surface electronic structure This content has been downloaded from IOPscience. Please scroll down to see the full text. 1982 Rep. Prog. Phys. 45 223 (http://iopscience.iop.org/0034-4885/45/3/001) View the table of contents for this issue, or go to the journal homepage for more Download details: IP Address: 152.17.136.208 This content was downloaded on 10/04/2015 at 02:48 Please note that terms and conditions apply. Rep. Prog. Phys., Vol. 45, 1982. Printed in Great Britain Surface electronic structure J E Inglesfield Science and Engineering Research Council, Daresbury Laboratory, Daresbury, Warrington WA4 4AD, UK Abstract The theory of the electronic structure of clean metal and semiconductor surfaces is reviewed, starting from an effective one-electron Schrodinger equation. Methods for solving the Schrodinger equation at surfaces are briefly described, and the effects of the surface on the electronic wavefunctions are discussed using simple models. The results of detailed calculations of the surface electronic structure of s-p bonded metals, transition metals and semiconductors are reviewed, with an emphasis on the effect of the local environment on the density of states. Properties like the work function and surface energy depend on the surface electronic structure, and their variation with material and surface is discussed; the surface energy contains an important contribution from the interaction between electrons, and this will be considered in some detail. The change in electronic structure compared with the bulk leads to changes in atomic structure, with surface reconstruction on semiconductor and some metal surfaces, and this is also discussed. -

Surface-State Bands on Silicon As Electron Systems in Reduced Dimensions at Atomic Scales
J. Phys.: Condens. Matter 12 (2000) R463–R495. Printed in the UK PII: S0953-8984(00)97483-6 REVIEW ARTICLE Surface-state bands on silicon as electron systems in reduced dimensions at atomic scales Shuji Hasegawa Department of Physics, Graduate School of Science, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan and Core Research for Evolutional Science and Technology, The Japan Science and Technology Corporation, Kawaguchi Centre Building, Hon-cho 4-1-8, Kawaguchi, Saitama 332-0012, Japan Received 25 May 2000 Abstract. When foreign atoms to a depth of around one atomic layer adsorb on silicon crystal surfaces, the adsorbates rearrange themselves by involving the substrate Si atoms; this results in peculiar periodic atomic arrangements, surface superstructures, in just the topmost surface layers. Then, characteristic electronic states are created there, which are sometimes quite different from the bulk electronic states in the interior of the crystal, leading to novel properties only at the surfaces. Here, surface superstructures are introduced that have two-dimensional or quasi- one-dimensional metallic electronic states on silicon surfaces. Sophisticated surface science techniques, e.g., scanning tunnelling microscopy, photoemission spectroscopy, electron-energy- loss spectroscopy, and microscopic four-point probes reveal characteristic phenomena such as phase transitions accompanying symmetry breakdown, electron standing waves, charge-density waves, sheet plasmons, and surface electronic transport, in which surface-state bands play main roles. These results show that surface superstructures on silicon provide fruitful platforms on which to investigate the physics of atomic-scale low-dimensional electron systems. 1. Introduction A silicon crystal has a diamond lattice, in which each Si atom has four ‘hands’ (valence electrons) with which to make covalent bonds with four neighbours in a tetrahedral arrangement. -

Shockley Surface States from First Principles
Shockley Surface States from First Principles Masterarbeit zur Erlangung des akademischen Grades eines Master of Science an der naturwissenschaftlichen Fakultät der Karl-Franzens-Universität Graz vorgelegt von Dario Wolf Knebl am Institut für Physik Fachbereich für theoretische Physik Betreuer: Ass.-Prof. Dr. Peter Puschnig Graz, 2013 In the first part of this thesis, a comprehensive ab-initio study within the frame- work of density functional theory (DFT) of all Shockley surface states of the (110), (100), and (111) facets of the face-centered cubic metals Al and Cu is performed. By comparing the bulk projected band structure with those of slab calculations, Shock- ley surface states are identified and a comprehensive analysis of their characteristic properties is conducted. Particular care is put on the convergence of calculated en- ergy positions and effective masses of surface states with respect to the number of metallic layers used in our repeated slab approach. In addition to band offsets and band dispersions, also computed surface energies, work functions, as well as typical decay lengths of surface states into the bulk and into the vacuum are compared among the investigated metals and facets and compared to available experimental data. Moreover, the DFT calculations are used to simulate angle-resolved photoe- mission (ARPES) intensities within the one-step model of photoemission. In the second part of the thesis, the prototypical organic/metal interface PTC- DA/Ag(110) is investigated. A careful convergence study on the projected density of states (pDOS) with respect to the number of atomic silver layers is performed. This highlights a parity dependence of the pDOS in the energy range of the lowest unoccupied molecule orbital (LUMO) on the number of silver layers. -

A Model Study of Surface State on Optical Bandgap of Silicon Nanowires
Science World Journal Vol 10 (No 4) 2015 www.scienceworldjournal.org ISSN 1597-6343 A MODEL STUDY OF SURFACE STATE ON OPTICAL BANDGAP OF SILICON NANOWIRES Full Length Research Article *1Aliyu I. Kabiru, 2Isah Mustapha, 3Abbas U. Dallatu and 3Danlami Abdulhadi *1Department of Physics, Faculty of Science, Kaduna State University, P.M.B 2339, Kaduna State, Nigeria 2Department of Applied Science, Kaduna Polytechnic, P.M.B 2021 Kaduna, Kaduna State, Nigeria 3Department of Science Laboratory Technology, School of applied Science, Nuhu Bamalli Polytechnic P.M.B 1061 Zaria, Kaduna state, Nigeria *Corresponding author Email: [email protected] ABSTRACT application in electronic and optoelectronic devices. In addition A theoretical approach is carried out to study the role of surface the correlation between the defects and the optical properties, it is state in silicon nanowires. The influences of size and surface also noticed that at nanoscale, the size not only directly impacts passivation on the bandgap energy and photoluminescence the defect contents but also the optical properties of materials spectra of silicon nanowires with diameter between 4 to 12nm are (Harrison 2005). Optical and electronic properties of SINWS are examined. It is observed that visible PL in silicon nanowires is due controlled by surface effect and quantum confinement. Recently, to quantum confinement and surface passivation. But the energy passivated surface Si nanostructures with hydrogen and oxygen recombination of electron and holes in the quantum confined become attractive due to enhanced luminescence observed in nanostructures is responsible for the visible PL. In this work, them. Geometry plays a crucial role in the enhanced surface models from quantum bandgap and photoluminescence intensity defect due to large surface-to-volume ratio (Ghoshal, 2005). -

Fermi Surfaces of Surface States on Si(111) + Ag, Au
Fermi Surfaces of Surface States on Si(111) + Ag, Au J. N. Crain, K. N. Altmann, Ch. Brombergera, and F. J. Himpsel Department of Physics, UW-Madison, 1150 University Ave., Madison, WI 53706, USA a Department of Physics, Philipps - University, Marburg, Germany Abstract Metallic surface states on semiconducting substrates provide an opportunity to study low-dimensional electrons decoupled from the bulk. Angle resolved photoemission is used to determine the Fermi surface, group velocity, and effective mass for surface states on Si(111)√3×√3-Ag, Si(111)√3×√3-Au, and Si(111)√21×√21-(Ag+Au). For Si(111)√3×√3-Ag the Fermi surface consists of small electron pockets populated by electrons from a few % excess Ag. For Si(111)√21×√21-(Ag+Au) the added Au forms a new, metallic band. The √21×√21 superlattice leads to an intricate surface umklapp pattern and to minigaps of 110 meV, giving an interaction potential of 55 meV for the √21×√21 superlattice. PACS: 73.20.At, 79.60.Jv, 73.25.+i 1. Introduction 2. Experiment Metallic surface states on semiconductors are special For preparing surface structures with optimum uniformity because they are the electronic equivalent of a two- and ordering we found the stoichiometry and the annealing dimensional electron gas in free space. Electronic states at the sequence to be the most critical parameters. First we will Fermi level EF are purely two-dimensional since there are no describe calibration of the optimum Au and Ag coverages by bulk states inside the gap of the semiconductor that they can low energy electron diffraction (LEED) and the optimum couple to. -

Spin- and Angle-Resolved Photoemission on the Topological Kondo Insulator Candidate
Spin- and angle-resolved photoemission on the topological Kondo Insulator candidate: SmB6 Nan Xu1,2, Hong Ding3,4 & Ming Shi1 1 Swiss Light Source, Paul Scherrer Institut, CH-5232 Villigen PSI, Switzerland 2 Institute of Condensed Matter Physics, École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland 3 Beijing National Laboratory for Condensed Matter Physics and Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China 4 Collaborative Innovation Center of Quantum Matter, Beijing, China Abstract Topological Kondo insulators are a new class of topological insulators in which metallic surface states protected by topological invariants reside in the bulk band gap at low temperatures. Unlike other three-dimensional topological insulators, a truly insulating bulk state, which is critical for potential applications in next-generation electronic devices, is guaranteed by many-body effects in the topological Kondo insulator. Furthermore, the system has strong electron correlations that can serve as a testbed for interacting topological theories. This topical review focuses on recent advances in the study of SmB6, the most promising candidate for a topological Kondo insulator, from the perspective of spin- and angle-resolved photoemission spectroscopy with highlights of some important transport results. Email: [email protected]; [email protected]; [email protected]. 42 Table of contents 1. Introduction. 1.1 Topological insulator 1.2 Topological Kondo insulator and predicted candidate: SmB6 2. Electronic structure of SmB6 probed by ARPES 2.1 Angle-resolved photoemission spectroscopy 2.2 Bulk hybridization gap and in-gap states in SmB6 2.3 Two-dimensionality and surface origin of the in-gap states 2.4 Surface reconstructions 2.5 Temperature evolution of electronic structure 3. -

A 5 Electronic Structure of Matter: Reduced Dimensions
A 5 Electronic structure of matter: Reduced dimensions Stefan Blugel¨ Institut fur¨ Festkorperforschung¨ Forschungszentrum Julich¨ Contents 1 Introduction 2 1.1Electronsinaperiodicpotential:singleelectronpicture............. 2 2Confined Electronic States 3 2.1 Jellium Model ................................... 4 2.2Quantum-WellStates............................... 6 3 Electronic Structure of Surfaces 9 3.1 Qualitative Discussion in One Dimension . ................. 10 3.2DescriptioninThreeDimensions......................... 12 3.3 Metallic Surfaces . ............................ 13 3.4 Semiconductor Surfaces . ............................ 19 4 Transition Metals in Reduced Dimensions: Magnetism 21 4.1RoleofCoordinationNumber.......................... 23 4.2Low-DimensionalMagnets............................ 27 A5.2 Stefan Bl¨ugel 1 Introduction In this lecture we discuss the consequences for the electronic structure of a crystalline solid if one or more dimensions of the system size are reduced, reduced to a level where measurable quantities are modified. This includes the change of the quantization conditions for an electron wave due to the presence of new boundary conditions which alters the eigenvalue spectrum and thus the transport and other properties of the solid. This is well-understood in terms of a sin- gle electron picture. With the reduced dimensions, the symmetries of the system are lowered, surfaces and interfaces move into the focus of attention and the appearance of new boundary conditions lead to additional states, called surface states and interface states. Typical for solids at reduced dimensions is also the reduction of the number of neighboring atoms or the reduction of the coordination number, respectively. This may request an rearrangement of atoms at the surface or interface of solids to find a new optimal bonding by lowering the energy. Typical examples are the surface reconstruction exhibited by many semiconductor surfaces rearranging their directional bonds or the formation of carbon nanotubes. -

Tight-Binding Electronic Band Structure and Surface States of Cu- OPENACCESS Chalcopyrite Semiconductors
Journal of Physics Communications PAPER • OPEN ACCESS Related content - Lattice structures and electronic properties Tight-binding electronic band structure and surface of WZ-CuInS2/WZ-CdS interface from first- principles calculations Hong-Xia Liu, Fu-Ling Tang, Hong-Tao states of Cu-chalcopyrite semiconductors Xue et al. - Electronic structure of (001) To cite this article: J E Castellanos-Aguila 2018 2 035021 et al J. Phys. Commun. (AlAs)t (GaAs)^(Al^s) m (GaAs)n superlattices L Fernandez-Alvarez, G Monsivais and V R Velasco View the article online for updates and enhancements. - Anisotropic features in the electronic structure of the two-dimensional transition metal trichalcogenide TiS3: electron doping and plasmons J A Silva-Guillen, E Canadell, P Ordejon et al. This content was downloaded from IP address 138.100.25.96 on 19/02/2020 at 10:00 IOP Publishing J. Phys. Commun. 2 (2018) 035021 https://doi.org/10.1088/2399-6528/aab10b Journal of Physics Communications PAPER (DCrossMar k Tight-binding electronic band structure and surface states of Cu- OPENACCESS chalcopyrite semiconductors 1 November 2017 J E Castellanos-Aguila1, P Palacios2'3, P Wahnon24 and J Arriaga1 REVISED 1 Instituto de Fisica, Benemerita Universidad Autonoma de Puebla, 18 SurySanClaudio, Edif. 110-A, Ciudad Univeristaria, 72570, Puebla, 8 February 2018 Mexico ACCEPTED FOR PUBLICATION 2 Instituto de Energia Solar, Universidad Politecnica de Madrid, 28040 Madrid, Spain 21 February2018 3 Dept. FAIAN, ETSI Aeronauticay del Espacio, Universidad Politecnica de Madrid, 28040, Madrid, Spain PUBLISHED 4 Dept. TFB, ETSI Telecomunicacion, Universidad Politecnica de Madrid, 28040 Madrid, Spain 9 March 2018 E-mail: [email protected] Original content from this Keywords: electronic band structure, surface states, intermediate band work may be used under the terms of the Creative Commons Attribution 3.0 licence. -
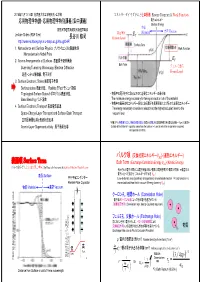
表面項 Surface Term
2018年11月12-14日 名古屋大学工学研究科・工学部 エネルギーダイヤグラムと仕事関数 Energy Diagram & Work Function 応用物理学特論・応用物理学特別講義(集中講義) 電子エネルギー Electron Energy 物質 東京大学理学系研究科物理学専攻 真空準位 Material 真空 Vacuum Lecture Slides (PDF files) EV 長谷川 修司 Vacuum Level http://www-surface.phys.s.u-tokyo.ac.jp/KougiOHP/ Surface Term 1.Nanoscience and Surface Physics ナノサイエンスと表面物理 Work Function Nanoscience in Nobel Prize 2.Atomic Arrangements at Surfaces 表面原子配列構造 EF Bulk Term Scanning Tunneling Microscopy, Electron Diffraction フェルミ準位 Fermi Level 走査トンネル顕微鏡、電子回折 3.Surface Electronic States 表面電子状態 Surface states 表面状態、 Rashba Effect ラシュバ効果 Topological Surface States トポロジカル表面状態、 • 物質中の電子を外に取り出すのに必要なエネルギーの最小値 Band Bending バンド湾曲 The minimum energy necessary for taking an electron out of the material • 物質中の最高占有エネルギー準位にある電子を真空準位に上げるのに必要なエネルギー 4.Surface Electronic Transport 表面電気伝導 The energy necessary to excite an electron at the highest occupied level to the Space-Charge-Layer Transport and Surface-State Transport vacuum level 空間電荷層伝導と表面状態伝導 「物質の外」:無限遠ではなく、物質の表面の直上(表面から鏡像力の影響を無視できる程度の距離 ~1 μm)の真空中 Atomic-Layer Superconductivity 原子層超伝導 Outside of the Material = a position away from the surface (~1 μm) at which the image force is ignored, not a position at infinite. バルク項 (交換相関エネルギーV )-(運動エネルギー) 表面項 XC Surface Term Bulk Term (Exchange-Correlation Energy VXC)-(Kinetic Energy) 電子の滲みだしと表面電気二重層 Spill out of electrons & Surface Electric Dipole Layer それぞれの電子の周りには電子密度の低い領域(正電荷を帯びた領域)が存在 ⇒真空中の 電子に比べて安定化 (エネルギーが下がる V ) 表面 Surface XC 平行平板コンデンサー Low-el-density area (positively charged area) around each electron ⇒ Each electron is Parallel-Plate Capacitor more stabilized than that in vacuum (Energy lowering VXC) 物質 Material 真空 Vacuum クーロン孔,相関ホール (Correlation Hole) 電子間のクーロン反発によって他の電子を遠ざけている + ー (相関相互作用) (Correlation Inter. due to Coulomb repulsion) + ー + ー フェルミ孔,交換ホール (Exchange Hole) 同じスピンを持つ電子どうしは,パウリの排他原理による + ー 物質 Potential) 交換相互作用による反発がはたらき他の電子を遠ざけている + ー RS - (Exchange Inter.