I. WEX.LXHSS, B.Sc. Proquest Number: 13850388
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biodegradation of 2-Chloro-4-Nitrophenol Via A
Min et al. AMB Expr (2018) 8:43 https://doi.org/10.1186/s13568-018-0574-7 ORIGINAL ARTICLE Open Access Biodegradation of 2‑chloro‑4‑nitrophenol via a hydroxyquinol pathway by a Gram‑negative bacterium, Cupriavidus sp. strain CNP‑8 Jun Min1, Jinpei Wang2, Weiwei Chen1 and Xiaoke Hu1* Abstract Cupriavidus sp. strain CNP-8 isolated from a pesticide-contaminated soil was able to utilize 2-chloro-4-nitrophenol (2C4NP) as a sole source of carbon, nitrogen and energy, together with the release of nitrite and chloride ions. It could degrade 2C4NP at temperatures from 20 to 40 °C and at pH values from 5 to 10, and degrade 2C4NP as high as 1.6 mM. Kinetics assay showed that biodegradation of 2C4NP followed Haldane substrate inhibition model, with the maximum specifc growth rate (μmax) of 0.148/h, half saturation constant (Ks) of 0.022 mM and substrate inhibi- tion constant (Ki) of 0.72 mM. Strain CNP-8 was proposed to degrade 2C4NP with hydroxyquinol (1,2,4-benzenetriol, BT) as the ring-cleavage substrate. The 2C4NP catabolic pathway in strain CNP-8 is signifcant from those reported in other Gram-negative 2C4NP utilizers. Enzymatic assay indicated that the monooxygenase initiating 2C4NP catabolism had diferent substrates specifcity compared with previously reported 2C4NP monooxygenations. Capillary assays showed that strain CNP-8 exhibited metabolism-dependent chemotactic response toward 2C4NP at the optimum concentration of 0.5 mM with a maximum chemotaxis index of 37.5. Furthermore, microcosm studies demonstrated that strain CNP-8, especially the pre-induced cells, could remove 2C4NP rapidly from the 2C4NP-contaminated soil. -

(10) Patent No.: US 8119385 B2
US008119385B2 (12) United States Patent (10) Patent No.: US 8,119,385 B2 Mathur et al. (45) Date of Patent: Feb. 21, 2012 (54) NUCLEICACIDS AND PROTEINS AND (52) U.S. Cl. ........................................ 435/212:530/350 METHODS FOR MAKING AND USING THEMI (58) Field of Classification Search ........................ None (75) Inventors: Eric J. Mathur, San Diego, CA (US); See application file for complete search history. Cathy Chang, San Diego, CA (US) (56) References Cited (73) Assignee: BP Corporation North America Inc., Houston, TX (US) OTHER PUBLICATIONS c Mount, Bioinformatics, Cold Spring Harbor Press, Cold Spring Har (*) Notice: Subject to any disclaimer, the term of this bor New York, 2001, pp. 382-393.* patent is extended or adjusted under 35 Spencer et al., “Whole-Genome Sequence Variation among Multiple U.S.C. 154(b) by 689 days. Isolates of Pseudomonas aeruginosa” J. Bacteriol. (2003) 185: 1316 1325. (21) Appl. No.: 11/817,403 Database Sequence GenBank Accession No. BZ569932 Dec. 17. 1-1. 2002. (22) PCT Fled: Mar. 3, 2006 Omiecinski et al., “Epoxide Hydrolase-Polymorphism and role in (86). PCT No.: PCT/US2OO6/OOT642 toxicology” Toxicol. Lett. (2000) 1.12: 365-370. S371 (c)(1), * cited by examiner (2), (4) Date: May 7, 2008 Primary Examiner — James Martinell (87) PCT Pub. No.: WO2006/096527 (74) Attorney, Agent, or Firm — Kalim S. Fuzail PCT Pub. Date: Sep. 14, 2006 (57) ABSTRACT (65) Prior Publication Data The invention provides polypeptides, including enzymes, structural proteins and binding proteins, polynucleotides US 201O/OO11456A1 Jan. 14, 2010 encoding these polypeptides, and methods of making and using these polynucleotides and polypeptides. -

Phase 11 Metabolism of Benzene Have to Be Identified
Phase 11 Metabolism of Benzene have to be identified. Application of single metabolites of benzene such as phenol Dieter Schrenk,1 Achim Orzechowski,1 (PH), catechol (CT), hydroquinone (HQ), and 1,2,4-trihydroxybenzene (THB) to Leslie R. Schwarz,2 Robert Snyder,3 Brian Burchell,4 rodents failed to reproduce the characteristic Magnus lngelman-Sundberg,5 and Karl Walter Bock1 toxic effects of benzene in the bone marrow (4,5). In several studies the possible syner- 1'nstitute of Toxicology, University of Tubingen, Tubingen, Germany; gistic action of certain metabolites of ben- 2GSF-lnstitute of Toxicology, Neuherberg/Munchen, Germany; zene on the bone marrow was investigated. 3Department of Pharmacology and Toxicology, EOHSI, Piscataway, It was shown that PH enhanced the conver- New Jersey; 4Department of Biochemical Medicine, University of sion of HQ into 1,4-benzoquinone cat- Dundee, Ninewells Hospital and Medical School, Dundee, United alyzed in vitro by myeloperoxidase (6,7), an Kingdom; 5Department of Physiological Chemistry, Karolinska Institute, enzyme present in abundance in the bone marrow (8). The electrophilic 1,4-benzo- Stockholm, Sweden quinone thus formed is able to bind to cel- The hepatic metabolism of benzene is thought to be a prerequisite for its bone marrow toxicity. lular proteins and DNA. Binding to critical However, the complete pattern of benzene metabolites formed in the liver and their role in bone proteins such as tubulin (9) or DNA poly- marrow toxicity are not fully understood. Therefore, benzene metabolism was studied in isolated merase-a (10) may play an important role rodent hepatocytes. Rat hepatocytes released benzene-1,2-dihydrodiol, hydroquinone (HQ), in benzene toxicity. -

Pugh and Raper
The Journal of Biochemistry , Vol. 35, No. 2. STUDIES ON TYROSINASE . VII. The action of the potatoe tyrosinase on trihydric phenols. BY HISASI SAITO. (Front the Biochemical Institute, Nagasaki Medical College . Director: Prof. Dr. T. Uchino.) (Received for publication, November 4, 1941) INTRODUCTION. Pugh and Raper (1927) pointed out that tyrosinase , being allowed to act on phenol, catechol, p-cresol and m-cresol in the presence of aniline, produces anilino compounds of o-quinones as oxidation products. They thus postulated the formation of ortho quinones from these phenols by the action of tyrosinase. The amount of oxygen, consumed in the enzymic oxidation of phenols, was, however, found by many investigators (Robinson and McCance, 1925; Pugh and Raper, 1927; Kawasaki, 1938; Sasaki, 1940, a) to be one atom more per molecule of substrates than that of corresponding to the formation of o-quinones. This probably indicates that o-quinones are not the final products of enzymic oxidation of phenolic substances. Wagreich and Nelson (1936) found that the amount of oxygen required for the oxidation of catechol by tyrosinase is two atoms and that the subsequent addition of aniline is accompanied by the absorption of another atom of oxygen with the formation of anilino-quinone. On the basis of this finding, they suggested that, in the case of catechol oxidation by tyrosinase, 4-hydroxy-l,2 - quinone is produced rather than o-benzoquinone as an iinternnediate product, and that hydroxyquinone thus formed then reacts withh aniline to produce dianilino quinone. On the other hand, Jackson (1939) examined the action of tyrosinase on cateehol as well as 1,2,4-tribydroxybenzene and came to the conclusion that 4-hydroxy 243 244 H. -

Amended Safety Assessment of Hydroquinone and P-Hydroxyanisole As Used in Cosmetics
Amended Safety Assessment of Hydroquinone and p-Hydroxyanisole as Used in Cosmetics Status: Draft Amended Report for Public Comment Release Date: November 15, 2013 Panel Meeting Date: December 9-10, 2013 The 2013 Cosmetic Ingredient Review Expert Panel members are: Chairman, Wilma F. Bergfeld, M.D., F.A.C.P.; Donald V. Belsito, M.D.; Curtis D. Klaassen, Ph.D.; Daniel C. Liebler, Ph.D.; Ronald A. Hill, Ph.D. James G. Marks, Jr., M.D.; Ronald C. Shank, Ph.D.; Thomas J. Slaga, Ph.D.; and Paul W. Snyder, D.V.M., Ph.D. The CIR Director is Lillian J. Gill, D.P.A. This report was prepared by Lillian C. Becker, Scientific Analyst/Writer. © Cosmetic Ingredient Review 1101 17th Street, NW, Suite 412 Washington, DC 20036-4702 ph 202.331.0651 fax 202.331.0088 cirinfo@cir- safety.org Distributed for Comment Only -- Do Not Cite or Quote Commitment & Credibility since 1976 MEMORANDOM To: CIR Expert Panel and Liaisons From: Lillian C. Becker, M.S. Scientific Analyst and Writer Date: November 15, 2013 Subject: Hydroquinone & p-Hydroxyanisole As Used In Nail Products in Cosmetics This is the Draft Report of hydroquinone and p-hydroxyanisole as used in nail products. In March, 2013, the Panel decided to review submitted data accompanying a request to amend the 2010 conclusion to include the use of nail polishes that require UV curing. This data, as well as relevant literature on the use of UV nail lamps, are included in this report. The Panel is to review the presented data and decide if there is sufficient information to come to a conclusion on the use of hydroquinone and p-hydroxyanisole in nail products that require UV curing. -

Amended Safety Assessment of Hydroquinone As Used in Cosmetics
Amended Safety Assessment of Hydroquinone as Used in Cosmetics Status: Draft Amended Report for Panel Review Release Date: February 21, 2014 Panel Meeting Date: March 17-18, 2014 The 2014 Cosmetic Ingredient Review Expert Panel members are: Chairman, Wilma F. Bergfeld, M.D., F.A.C.P.; Donald V. Belsito, M.D.; Curtis D. Klaassen, Ph.D.; Daniel C. Liebler, Ph.D.; Ronald A. Hill, Ph.D.; James G. Marks, Jr., M.D.; Ronald C. Shank, Ph.D.; Thomas J. Slaga, Ph.D.; and Paul W. Snyder, D.V.M., Ph.D. The CIR Director is Lillian J. Gill, D.P.A. This report was prepared by Lillian C. Becker, Scientific Analyst/Writer. © Cosmetic Ingredient Review 1620 L Street, NW, Suite 1200 Washington, DC 20036-4702 ph 202.331.0651 fax 202.331.0088 [email protected] i Commitment & Credibility since 1976 MEMORANDOM To: CIR Expert Panel and Liaisons From: Lillian C. Becker, M.S. Scientific Analyst and Writer Date: February 21, 2014 Subject: Hydroquinone As Used In in Cosmetics In December 2013, the Panel tabled the Draft Amended Report of hydroquinone and p- hydroxyanisole as used in nail products for the purpose of collecting more data on the use of UV nail lamps. The papers that were requested have been sent to you by email and are summarized in the appropriate section of the report. A paper that Dr. Liebler asked to review (Hansch et al. 2000) is also included in the email. The previous safety assessments on these ingredients are also included in case more information than what is provided in the summaries is needed. -
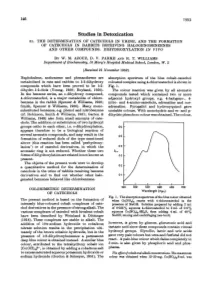
Studies in Detoxication 51
146 1953 Studies in Detoxication 51. THE DETERMINATION OF CATECHOLS IN URINE, AND THE FORMATION OF CATECHOLS IN RABBITS RECEIVING HALOGENOBENZENES AND OTHER COMPOUNDS. DIHYDROXYLATION IN VIVO BY W. M. AZOUZ, D. V. PARKE AND R. T. WILLIAMS Department of Biochemistry, St Mary's Hospital Medical School, London, W. 2 (Received 21 November 1952) Naphthalene, anthracene and phenanthrene are absorption spectrum of the blue cobalt-catechol metabolized in rats and rabbits to 1:2-dihydroxy coloured complex using 4-chlorocatechol is shown in compounds which have been proved to be 1:2- Fig. 1. dihydro-1:2-diols (Young, 1950; Boyland, 1950). The colour reaction was given by all aromatic In the benzene series, an o-dihydroxy compound, compounds tested which contained two or more 4-chlorocatechol, is a major metabolite of chloro- adjacent hydroxyl groups, e.g. 4-halogeno-, 4- benzene in the rabbit (Spencer & Williams, 1950; nitro- and 4-amino-catechols, adrenaline and nor- Smith, Spencer & Williams, 1950). Many mono- adrenaline. Pyrogallol and hydroxyquinol gave substituted benzenes, e.g. phenol and nitrobenzene unstable colours. With monohydric and m- and p- (cf. Robinson, Smith & Williams, 1951; Garton & dihydric phenols no colour was obtained. The colour, Williams, 1949) also form small amounts of cate- chols. The addition or substitution of two hydroxyl groups ortho to each other, i.e. o-dihydroxylation, 0-6 appears therefore to be a biological reaction of several aromatic compounds, and may result in the 0o5 formation of reduced diols of the type mentioned [ above (this reaction has been called 'perhydroxy- lation') or of catechol derivatives, in which the 0-4 aromatic ring is not reduced. -

Metabolic Pathways for Degradation of Aromatic Hydrocarbons by Bacteria
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/285049749 Metabolic Pathways for Degradation of Aromatic Hydrocarbons by Bacteria Article in Reviews of environmental contamination and toxicology · November 2015 DOI: 10.1007/978-3-319-23573-8_5 CITATIONS READS 18 2,127 5 authors, including: Guillermo Ladino-Orjuela Eleni Gomes Centro Universitário de Votuporanga São Paulo State University 7 PUBLICATIONS 31 CITATIONS 216 PUBLICATIONS 3,373 CITATIONS SEE PROFILE SEE PROFILE Roberto Da Silva John Parsons São Paulo State University University of Amsterdam 217 PUBLICATIONS 3,443 CITATIONS 165 PUBLICATIONS 2,874 CITATIONS SEE PROFILE SEE PROFILE Some of the authors of this publication are also working on these related projects: ENFIRO View project Application of enzymes in food science View project All content following this page was uploaded by Guillermo Ladino-Orjuela on 19 November 2017. The user has requested enhancement of the downloaded file. Metabolic Pathways for Degradation of Aromatic Hydrocarbons by Bacteria Guillermo Ladino-Orjuela , Eleni Gomes , Roberto da Silva , Christopher Salt , and John R. Parsons Contents 1 Introduction ....................................................................................................................... 105 2 Biodegradation of Aromatic Compounds ......................................................................... 107 2.1 Aromatic Hydrocarbon Biodegradation Under Aerobic Conditions ....................... 108 2.2 Aromatic Hydrocarbon -

Cloning of Two Gene Clusters Involved in the Catabolism of 2,4-Dinitrophenol by Paraburkholderia Sp
bioRxiv preprint doi: https://doi.org/10.1101/749879; this version posted August 29, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Cloning of Two Gene Clusters Involved in the Catabolism of 2,4-Dinitrophenol by Paraburkholderia sp. 2 Strain KU-46 and Characterization of the Initial DnpAB Enzymes and a Two-Component 3 Monooxygenase DnpC1C2 4 5 Taisei Yamamoto,a Yaxuan Liu,a Nozomi Kohaya,a Yoshie Hasegawa,a Peter C.K. Lau,b Hiroaki Iwakia# 6 7 aDepartment of Life Science & Biotechnology, Kansai University, Suita, Osaka, Japan 8 bDepartment of Microbiology and Immunology, McGill University, Montréal, Quebec, Canada 9 10 11 Running Head: Unique Pathway of 2,4-Dinitrophenol Catabolism 12 #Address correspondence to Hiroaki Iwaki, [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/749879; this version posted August 29, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 13 Abstract 14 Besides an industrial pollutant, 2,4-dinitrophenol (DNP) has been used illegally as a weight loss drug that 15 had claimed human lives. Little is known about the metabolism of DNP, particularly among 16 Gram-negative bacteria. In this study, two non-contiguous genetic loci of Paraburkholderia (formerly 17 Burkholderia) sp. strain KU-46 genome were identified and four key initial genes (dnpA, dnpB, and 18 dnpC1C2) were characterized to provide molecular and biochemical evidence for the degradation of DNP 19 via the formation of 4-nitrophenol (NP), a pathway that is unique among DNP utilizing bacteria. -

(12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur Et Al
US 20150240226A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur et al. (43) Pub. Date: Aug. 27, 2015 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/16 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/02 (2006.01) CI2N 9/78 (2006.01) (71) Applicant: BP Corporation North America Inc., CI2N 9/12 (2006.01) Naperville, IL (US) CI2N 9/24 (2006.01) CI2O 1/02 (2006.01) (72) Inventors: Eric J. Mathur, San Diego, CA (US); CI2N 9/42 (2006.01) Cathy Chang, San Marcos, CA (US) (52) U.S. Cl. CPC. CI2N 9/88 (2013.01); C12O 1/02 (2013.01); (21) Appl. No.: 14/630,006 CI2O I/04 (2013.01): CI2N 9/80 (2013.01); CI2N 9/241.1 (2013.01); C12N 9/0065 (22) Filed: Feb. 24, 2015 (2013.01); C12N 9/2437 (2013.01); C12N 9/14 Related U.S. Application Data (2013.01); C12N 9/16 (2013.01); C12N 9/0061 (2013.01); C12N 9/78 (2013.01); C12N 9/0071 (62) Division of application No. 13/400,365, filed on Feb. (2013.01); C12N 9/1241 (2013.01): CI2N 20, 2012, now Pat. No. 8,962,800, which is a division 9/2482 (2013.01); C07K 2/00 (2013.01); C12Y of application No. 1 1/817,403, filed on May 7, 2008, 305/01004 (2013.01); C12Y 1 1 1/01016 now Pat. No. 8,119,385, filed as application No. PCT/ (2013.01); C12Y302/01004 (2013.01); C12Y US2006/007642 on Mar. 3, 2006. -
Purification of Hydroxyquinol 1,2-Dioxygenase And
APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Nov. 1996, p. 4276–4279 Vol. 62, No. 11 0099-2240/96/$04.0010 Copyright q 1996, American Society for Microbiology Purification of Hydroxyquinol 1,2-Dioxygenase and Maleylacetate Reductase: the Lower Pathway of 2,4,5-Trichlorophenoxyacetic Acid Metabolism by Burkholderia cepacia AC1100 DAYNA L. DAUBARAS, KATSUHIKO SAIDO,† AND A. M. CHAKRABARTY* Department of Microbiology and Immunology, College of Medicine, University of Illinois at Chicago, Chicago, Illinois 60612 Received 10 June 1996/Accepted 26 August 1996 The enzyme hydroxyquinol 1,2-dioxygenase, which catalyzes ortho cleavage of hydroxyquinol (1,2,4-trihy- droxybenzene) to produce maleylacetate, was purified from Escherichia coli cells containing the tftH gene from Burkholderia cepacia AC1100. Reduction of the double bond in maleylacetate is catalyzed by the enzyme maleylacetate reductase, which was also purified from E. coli cells, these cells containing the tftE gene from B. cepacia AC1100. The two enzymes together catalyzed the conversion of hydroxyquinol to 3-oxoadipate. The purified hydroxyquinol 1,2-dioxygenase was specific for hydroxyquinol and was not able to use catechol, tetrahydroxybenzene, 6-chlorohydroxyquinol, or 5-chlorohydroxyquinol as its substrate. The native molecular mass of hydroxyquinol 1,2-dioxygenase was 68 kDa, and the subunit size of the protein was 36 kDa, suggesting a dimeric protein of identical subunits. Aerobic metabolism of chloroaromatic compounds occurs of 2,4,5-T degradation, involving the metabolism of 5-chloro- through two pathways. Generally, simple chlorinated aromatic 1,2,4-trihydroxybenzene, the genes essential for the comple- compounds containing one or two chlorine substituents are mentation of the mutant B. -

Toxic Substances — Focus on Children Developing a Canadian List of Substances of Concern to Children’S Health
Toxic Substances — Focus on Children Developing a Canadian List of Substances of Concern to Children’s Health Toxic Substances — Focus on Children Developing a Canadian List of Substances of Concern to Children’s Health June 2004 Prepared by: Kathleen Cooper Senior Researcher, Canadian Environmental Law Association Prepared for: Canadian Environmental Law Association and Pollution Probe CELA and Pollution Probe 1 Toxic Substances — Focus on Children Developing a Canadian List of Substances of Concern to Children’s Health The Canadian Environmental Law Pollution Probe is a non-profit charitable Association (CELA) is a non-profit, public organization that works in partnership with interest organization established in 1970 to all sectors of society to protect health by use existing laws to protect the environment promoting clean air and clean water. and to advocate environmental law reforms. Pollution Probe was established in 1969 at It is also a free legal advisory clinic for the the University of Toronto. Early issues tackled public, and will act at hearings and in courts by Pollution Probe included urging the on behalf of citizens or citizens’ groups who Canadian government to ban DDT for almost are otherwise unable to afford legal all uses, and campaigning for the clean-up of assistance. Funded by Legal Aid Ontario, the Don River in Toronto. We encouraged CELA is one of 79 community legal clinics curbside recycling in 140 Ontario located across Ontario, 15 of which offer communities and supported the development services in specialized areas of the law. of the Blue Box programme. CELA’s Objectives: Since the 1990s, Pollution Probe has focused • To provide equitable access to justice to its programmes on issues related to air those otherwise unable to afford pollution, water pollution, climate change representation for their environmental and human health, including a major problems; programme to remove human sources of • To advocate for comprehensive laws, mercury from the environment.