Alteration of Striatal Dopaminergic Neurotransmission in a Mouse Model of DYT11 Myoclonus-Dystonia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cover Petra B
UvA-DARE (Digital Academic Repository) The role of Polycomb group proteins throughout development : f(l)avoring repression van der Stoop, P.M. Link to publication Citation for published version (APA): van der Stoop, P. M. (2009). The role of Polycomb group proteins throughout development : f(l)avoring repression. Amsterdam: Nederlands Kanker Instituut - Antoni Van Leeuwenhoekziekenhuis. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (http://dare.uva.nl) Download date: 27 Oct 2019 Chapter 4 Ubiquitin E3 Ligase Ring1b/Rnf2 of Polycomb Repressive Complex 1 Contributes to Stable Maintenance of Mouse Embryonic Stem Cells Petra van der Stoop*, Erwin A. Boutsma*, Danielle Hulsman, Sonja Noback, Mike Heimerikx, Ron M. Kerkhoven, J. Willem Voncken, Lodewyk F.A. Wessels, Maarten van Lohuizen * These authors contributed equally to this work Adapted from: PLoS ONE (2008) 3(5): e2235 Ring1b regulates ES cell fate Ubiquitin E3 Ligase Ring1b/Rnf2 of Polycomb Repressive Complex 1 Contributes to Stable Maintenance of Mouse Embryonic Stem Cells Petra van der Stoop1*, Erwin A. -

SGCE Rabbit Pab
Leader in Biomolecular Solutions for Life Science SGCE Rabbit pAb Catalog No.: A5330 Basic Information Background Catalog No. This gene encodes the epsilon member of the sarcoglycan family. Sarcoglycans are A5330 transmembrane proteins that are components of the dystrophin-glycoprotein complex, which link the actin cytoskeleton to the extracellular matrix. Unlike other family Observed MW members which are predominantly expressed in striated muscle, the epsilon 55kDa sarcoglycan is more broadly expressed. Mutations in this gene are associated with myoclonus-dystonia syndrome. This gene is imprinted, with preferential expression from Calculated MW the paternal allele. Alternatively spliced transcript variants encoding different isoforms 49kDa/51kDa/52kDa have been found for this gene. A pseudogene associated with this gene is located on chromosome 2. Category Primary antibody Applications WB,IHC,IF Cross-Reactivity Human, Mouse Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 8910 O43556 IHC 1:50 - 1:200 Immunogen 1:50 - 1:200 IF Recombinant fusion protein containing a sequence corresponding to amino acids 1-317 of human SGCE (NP_003910.1). Synonyms SGCE;DYT11;ESG;epsilon-SG Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3. Validation Data Western blot analysis of extracts of various cell lines, using SGCE antibody (A5330) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (AS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (RM00020). -

2021 Code Changes Reference Guide
Boston University Medical Group 2021 CPT Code Changes Reference Guide Page 1 of 51 Background Current Procedural Terminology (CPT) was created by the American Medical Association (AMA) in 1966. It is designed to be a means of effective and dependable communication among physicians, patients, and third-party payers. CPT provides a uniform coding scheme that accurately describes medical, surgical, and diagnostic services. CPT is used for public and private reimbursement systems; development of guidelines for medical care review; as a basis for local, regional, and national utilization comparisons; and medical education and research. CPT Category I codes describe procedures and services that are consistent with contemporary medical practice. Category I codes are five-digit numeric codes. CPT Category II codes facilitate data collection for certain services and test results that contribute to positive health outcomes and quality patient care. These codes are optional and used for performance management. They are alphanumeric five-digit codes with the alpha character F in the last position. CPT Category III codes represent emerging technologies. They are alphanumeric five-digit codes with the alpha character T in the last position. The CPT Editorial Panel, appointed by the AMA Board of Trustees, is responsible for maintaining and updating the CPT code set. Purpose The AMA makes annual updates to the CPT code set, effective January 1. These updates include deleted codes, revised codes, and new codes. It’s important for providers to understand the code changes and the impact those changes will have to systems, workflow, reimbursement, and RVUs. This document is meant to assist you with this by providing a summary of the changes; a detailed breakdown of this year’s CPT changes by specialty, and HCPCS Updates for your reference. -

Supplemental Table 1. List of Candidate Gene Filters Used in the Analysis of Exome Sequencing. MYOPATHY NEUROPATHY MND ABHD5
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Supplemental table 1. List of candidate gene filters used in the analysis of exome sequencing. MYOPATHY NEUROPATHY MND ABHD5 AAAS AAAS ACADL AARS1 AARS1 ACADM ABCA1 AGT ACADS ABCD1 ALAD ACADVL ABHD12 ALS2 ACTA1 ADCY6 ANG ADSSL1 AFG3L2 APEX1 AGL AIFM1 APOE AGPAT2 AMACR AR AGRN ANG ASAH1 AIRE AP1S1 ATM ALDOA APOA1 ATP7A ALG14 APTX ATXN2 ALG2 ARHGEF10 ATXN3 ALG3 ARL6IP1 B4GALT6 ANKRD2 ARSA BCL11B ANO5 ASAH1 BCL6 ASCC1 ATL1 BICD2 ATGL ATL3 BSCL2 ATP2A1 ATM C19orf12 ATRN ATXN1 C9orf72 B3GALNT2 ATXN10 CCS B3GNT2 ATXN2 CDH13 BAG3 ATXN3 CDH22 BIN1 ATXN7 CHCHD10 BSCL2 B2M CHMP2B BVES B4GALNT1 CNTF CACNA1S BAG3 CNTN4 CAPN3 BCKDHB CNTN6 CASQ1 BSCL2 CRIM1 CAV1 C12orf65 CRYM CAV3 C9orf72 CSNK1G3 CAVIN1 CLP1 CST3 CCDC78 CNTNAP1 CUL4B CDKN1C COX10 CYP2D6 CFL2 COX6A1 DAO Grunseich C, et al. J Neurol Neurosurg Psychiatry 2021;0:1–11. doi: 10.1136/jnnp-2020-325437 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry CHAT CPOX DCAF15 CHCHD10 CRYAB DCTN1 CHD7 CTDP1 DIAPH3 CHKB CTSA DISC1 CHN1 CYP27A1 DNAJB2 CHRM3 DARS2 DOC2B CHRNA1 DDHD1 DPP6 CHRNB1 DGUOK DYNC1H1 CHRND DHH EFEMP1 CHRNE DHTKD1 ELP3 CIDEC DMD EPHA4 CLCN1 DNAJB2 EWSR1 CLN3 DNAJC3 EXOSC3 CNBP DNM2 FBLN5 CNTN1 DYNC1H1 FBXO38 COA3 EGR2 FEZF2 COL12A1 EMD FGGY COL13A1 ERCC6 FIG4 COL6A ERCC8 FUS COL6A1 FAH GARS1 COL6A2 FAM126A GBE1 COL6A3 FBLN5 GMPPA COL9A3 FGD4 GRB14 COLQ FGF14 GRN COX10 FIG4 HEXA COX15 FLNC HFE CPT2 FLRT1 HINT1 CRAT FLVCR1 HSPB1 CRPPA FMR1 HSPB3 CRYAB FUS HSPB8 CTNS FXN IGHMBP2 DAG1 GALC ITPR2 DECR1 GAN KDR DES GARS1 KIFAP3 DGUOK GBA2 KLHL9 DIH1 GBE1 LAMA2 DMD GDAP1 LAS1L DMPK GJB1 LIF DNAJB6 GJB3 LIPC DNAJC19 GLA LOX Grunseich C, et al. -

Neuromuscular Disorders
Neuromuscular Disorders neuromuscular disorders panel, as well as an expanded 78-gene panel for the following conditions depending on the EGL offers a 46-gene neuromuscular disorders panel, as well as an expanded 78-gene panel, depending on the specificity of a patient's phenotype. Other phenotype-specific panels are available for limb-girdle muscular dystrophy (34 Neuromuscular Disorders Genes Included on the Expanded Neuromuscular Disorders Panel* ACTA1 CHRNA1 DAG1 FLNC LMNA PLEKHG5 SCN4A TNNI2 AMPD1 CHRNB1 DES GAA MTM1 PMM2 SEPN1 TNNT1 ANO5 CHRND DMD GLE1 MTMR14 POMGNT1 SGCA TPM2 BAG3 CHRNE DNM2 GNE MUSK POMT1 SGCB TPM3 BIN1 CHRNG DOK7 IGHMBP2 MYH2 POMT2 SGCD TRIM32 BSCL2 COL6A1 DYSF ISPD MYH7 PTRF SGCE TTN CAPN3 COL6A2 EMD ITGA7 MYOT PYGM SGCG VCP CAV3 COL6A3 FHL1 LAMA2 NEB RAPSN SIL1 VRK1 CFL2 COLQ FKRP LARGE PABPN1 RYR1 SYNE1 CHAT CRYAB FKTN LDB3 PLEC RYR2 TCAP *Bolded genes are also found on the 46-gene neuromuscular disorders panel. Please note that deletion/duplication analysis is not completed for all genes in the panel. Some genes on this panel are associated with additional phenotypes. All genes on the next generation sequencing panel may be ordered separately. Genes included on panels are subject to change. Test Code Test Name CPT®** Code(s) 81400 (x1), 81401 (x1), 81404 (x1), MNEU1 Neuromuscular Disorders: Sequencing Panel 81405 (x1), 81406 (x1), 81407 (x1), 81408 (x1) 81161 (x1), 81404 (x1), 81405 (x1), DNEU1 Neuromuscular Disorders: Deletion/Duplication Panel 81406 (x1), 81408 (x1) MM360 Expanded Neuromuscular Disorders: -

Genetic Testing Medical Policy – Genetics
Genetic Testing Medical Policy – Genetics Please complete all appropriate questions fully. Suggested medical record documentation: • Current History & Physical • Progress Notes • Family Genetic History • Genetic Counseling Evaluation *Failure to include suggested medical record documentation may result in delay or possible denial of request. PATIENT INFORMATION Name: Member ID: Group ID: PROCEDURE INFORMATION Genetic Counseling performed: c Yes c No **Please check the requested analyte(s), identify number of units requested, and provide indication/rationale for testing. 81400 Molecular Pathology Level 1 Units _____ c ACADM (acyl-CoA dehydrogenase, C-4 to C-12 straight chain, MCAD) (e.g., medium chain acyl dehydrogenase deficiency), K304E variant _____ c ACE (angiotensin converting enzyme) (e.g., hereditary blood pressure regulation), insertion/deletion variant _____ c AGTR1 (angiotensin II receptor, type 1) (e.g., essential hypertension), 1166A>C variant _____ c BCKDHA (branched chain keto acid dehydrogenase E1, alpha polypeptide) (e.g., maple syrup urine disease, type 1A), Y438N variant _____ c CCR5 (chemokine C-C motif receptor 5) (e.g., HIV resistance), 32-bp deletion mutation/794 825del32 deletion _____ c CLRN1 (clarin 1) (e.g., Usher syndrome, type 3), N48K variant _____ c DPYD (dihydropyrimidine dehydrogenase) (e.g., 5-fluorouracil/5-FU and capecitabine drug metabolism), IVS14+1G>A variant _____ c F13B (coagulation factor XIII, B polypeptide) (e.g., hereditary hypercoagulability), V34L variant _____ c F2 (coagulation factor 2) (e.g., -

The Effect of Mechanical Force on Gene Expression of Human Bladder Smooth Muscle Cells" (2012)
Philadelphia College of Osteopathic Medicine DigitalCommons@PCOM PCOM Biomedical Studies Student Scholarship Student Dissertations, Theses and Papers 6-2012 The ffecE t of Mechanical Force on Gene Expression of Human Bladder Smooth Muscle Cells Christopher A. Callan Philadelphia College of Osteopathic Medicine, [email protected] Follow this and additional works at: http://digitalcommons.pcom.edu/biomed Part of the Molecular Genetics Commons, and the Urogenital System Commons Recommended Citation Callan, Christopher A., "The Effect of Mechanical Force on Gene Expression of Human Bladder Smooth Muscle Cells" (2012). PCOM Biomedical Studies Student Scholarship. Paper 36. This Thesis is brought to you for free and open access by the Student Dissertations, Theses and Papers at DigitalCommons@PCOM. It has been accepted for inclusion in PCOM Biomedical Studies Student Scholarship by an authorized administrator of DigitalCommons@PCOM. For more information, please contact [email protected]. PHILADELPHIA COLLEGE OF OSTEOPATHIC MEDICINE Philadelphia, Pennsylvania The Effect Of Mechanical Force on Gene Expression of Human Bladder Smooth Muscle Cells A thesis submitted in partial fulfillnment of the requirements for the degree of MASTER OF BIOMEDICAL SCIENCE by Christopher A. Callan June 2012 We approve the thesis of Christopher A. Callan _______________________________________________________________________ Edward J. Macarak, Ph.D. Date Chairman; Department of Anatomy and Cell Biology University of Pennsylvania School of Dental Medicine Thesis -

ISPD Mutations Account for a Small Proportion of Italian Limb Girdle Muscular Dystrophy Cases
Magri et al. BMC Neurology DOI 10.1186/s12883-015-0428-8 RESEARCH ARTICLE Open Access ISPD mutations account for a small proportion of Italian Limb Girdle Muscular Dystrophy cases Francesca Magri1†, Irene Colombo2†, Roberto Del Bo1, Stefano Previtali3, Roberta Brusa1, Patrizia Ciscato2, Marina Scarlato3, Dario Ronchi1, Maria Grazia D’Angelo4, Stefania Corti1, Maurizio Moggio2, Nereo Bresolin1 and Giacomo Pietro Comi1* Abstract Background: Limb Girdle Muscular Dystrophy (LGMD), caused by defective α-dystroglycan (α-DG) glycosylation, was recently associated with mutations in Isoprenoid synthase domain-containing (ISPD) and GDP-mannose pyrophosphorylase B (GMPPB) genes. The frequency of ISPD and GMPPB gene mutations in the LGMD population is unknown. Methods: We investigated the contributions of ISPD and GMPPB genes in a cohort of 174 Italian patients with LGMD, including 140 independent probands. Forty-one patients (39 probands) from this cohort had not been genetically diagnosed. The contributions of ISPD and GMPPB were estimated by sequential α-DG immunohistochemistry (IHC) and mutation screening in patients with documented α-DG defect, or by direct DNA sequencing of both genes when muscle tissue was unavailable. Results: We performed α-DG IHC in 27/39 undiagnosed probands: 24 subjects had normal α-DG expression, two had a partial deficiency, and one exhibited a complete absence of signal. Direct sequencing of ISPD and GMPPB revealed two heterozygous ISPD mutations in the individual who lacked α-DG IHC signal: c.836-5 T > G (which led to the deletion of exon 6 and the production of an out-of-frame transcript) and c.676 T > C (p.Tyr226His). -

HALOPLEX DISEASE RESEARCH PANELS Premium Performance with Content You Can Trust
NGS TARGET ENRICHMENT HALOPLEX DISEASE RESEARCH PANELS Premium Performance with Content You Can Trust The ability to detect sequence level variations through next generation Benefits sequencing in a fast, cost effective application has revolutionized the field of genetics. In order to harness the power of next generation sequencing for specific Confidence in Genomic Content disease states, it is critical to target these specific genomic regions. Developed • Comprehensive content tailored to in conjunction with clinical research industry leaders, Agilent Technologies offers specific disease research HaloPlex next generation sequencing target enrichment panels for researching specific genetic disorders. • Developed in conjunction with top clinical research leaders From Sample to Result in Less than 2 Days Ease of Use From Sample to Analysis 1 Order HaloPlex kit • Simple, automatable protocol with Ready to Order or Made to Order no library preparation Cancer Arrhythmia Cardiomyopathy Connective Tissue Disorder • SureCall software for intuitive Noonan Syndrome variant analysis and report formats ICCG X Chromosome Available on Illumina MiSeq Results You Can Trust and Ion PGM • Multiple amplicon coverage of targets providing better coverage, 2 less PCR artifacts and more Prepare Samples and Sequence accurate mutation calls • Premium performance including high sensitivity, specificity, and mutation detection 3 Analyze data, print/export results SureCall See Deeper. Reach Further. CATALOG PANELS ClearSeq AML HaloPlex Cancer HaloPlex Cardiomyopathy The ClearSeq AML, designed in collabora- HaloPlex Cancer is a comprehensive next HaloPlex Cardiomyopathy is a next tion with Dr. Robert Ohgami and Dr. Daniel generation sequencing target enrichment generation sequencing target enrichment Arber at Stanford University, targets 48 panel designed specifically for genetic panel designed specifically for inherited selected exons in 20 genes found to be anomalies in known cancer hotspots. -

Quantitative Proteomic Comparison of Myofibroblasts Derived from Bone
www.nature.com/scientificreports OPEN Quantitative proteomic comparison of myofbroblasts derived from bone marrow and cornea Paramananda Saikia1,4, Jack S. Crabb1,2,4, Luciana L. Dibbin1, Madison J. Juszczak1, Belinda Willard2, Geeng‑Fu Jang1,2, Thomas Michael Shiju1, John W. Crabb1,2,3* & Steven E. Wilson1,3* Myofbroblasts are fbroblastic cells that function in wound healing, tissue repair and fbrosis, and arise from bone marrow (BM)‑derived fbrocytes and a variety of local progenitor cells. In the cornea, myofbroblasts are derived primarily from stromal keratocytes and from BM‑derived fbrocytes after epithelial‑stromal and endothelial‑stromal injuries. Quantitative proteomic comparison of mature alpha‑smooth muscle actin (α‑SMA)+ myofbroblasts (verifed by immunocytochemistry for vimentin, α‑SMA, desmin, and vinculin) generated from rabbit corneal fbroblasts treated with transforming growth factor (TGF) beta‑1 or generated directly from cultured BM treated with TGF beta‑1 was pursued for insights into possible functional diferences. Paired cornea‑derived and BM‑derived α‑SMA+ myofbroblast primary cultures were generated from four New Zealand white rabbits and confrmed to be myofbroblasts by immunocytochemistry. Paired cornea‑ and BM‑derived myofbroblast specimens from each rabbit were analyzed by LC MS/MS iTRAQ technology using an Orbitrap Fusion Lumos Tribrid mass spectrometer, the Mascot search engine, the weighted average quantifcation method and the UniProt rabbit and human databases. From 2329 proteins quantifed with ≥ 2 unique peptides from ≥ 3 rabbits, a total of 673 diferentially expressed (DE) proteins were identifed. Bioinformatic analysis of DE proteins with Ingenuity Pathway Analysis implicate progenitor‑dependent functional diferences in myofbroblasts that could impact tissue development. -
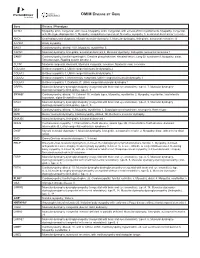
Please Find the Updated Gene List Here
OMIM DISEASE BY GENE Gene Disease / Phenotype ACTA1 Myopathy, actin, congenital, with cores, Myopathy, actin, congenital, with excess of thin myofilaments, Myopathy, congenital, with fiber-type disproportion 1, Myopathy, scapulohumeroperoneal, Nemaline myopathy 3, autosomal dominant or recessive ANO5 Gnathodiaphyseal dysplasia, Miyoshi muscular dystrophy 3, Muscular dystrophy, limb-girdle, autosomal recessive 12 ATP2A1 Brody myopathy BAG3 Cardiomyopathy, dilated, 1HH, Myopathy, myofibrillar, 6 CAPN3 Muscular dystrophy, limb-girdle, autosomal dominant 4, Muscular dystrophy, limb-girdle, autosomal recessive 1 CAV3* Cardiomyopathy, familial hypertrophic, Creatine phosphokinase, elevated serum, Long QT syndrome 9, Myopathy, distal, Tateyama type, Rippling muscle disease 2 CLCN1 Myotonia congenita, dominant, Myotonia congenita, recessive, Myotonia levior, recessive COL12A1 Bethlem myopathy 2, Ullrich congenital muscular dystrophy 2 COL6A1 Bethlem myopathy 1, Ullrich congenital muscular dystrophy 1 COL6A2 Bethlem myopathy 1, Myosclerosis, congenital, Ullrich congenital muscular dystrophy 1 COL6A3 Bethlem myopathy 1, Dystonia 27, Ullrich congenital muscular dystrophy 1 CRPPA Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 7, Muscular dystrophy- dystroglycanopathy (limb-girdle), type C, 7 CRYAB* Cardiomyopathy, dilated, 1II, Cataract 16, multiple types, Myopathy, myofibrillar, 2, Myopathy, myofibrillar, fatal infantile hypertonic, alpha-B crystallin-related DAG1 Muscular dystrophy-dystroglycanopathy (congenital -

Proteomic Profiling of the Interface Between the Stomach Wall and The
Proteomics of stomach wall and pancreas in dystrophinopathy Eur J Transl Myol 31 (1): 9627, 2021 doi: 10.4081/ejtm.2021.9627 Proteomic profiling of the interface between the stomach wall and the pancreas in dystrophinopathy Paul Dowling (1,2), Stephen Gargan (1,2), Margit Zweyer (3), Hemmen Sabir (3), Michael Henry (4), Paula Meleady (4), Dieter Swandulla (5), Kay Ohlendieck (1,2) (1) Department of Biology, Maynooth University, National University of Ireland, Maynooth, Co. Kildare, Ireland; (2) Kathleen Lonsdale Institute for Human Health Research, Maynooth University, Maynooth, Co. Kildare, Ireland, (3) Department of Neonatology and Paediatric Intensive Care, Children’s Hospital, University of Bonn, Bonn, Germany; (4) National Institute for Cellular Biotechnology, Dublin City University, Dublin 9, Ireland; (5) Institute of Physiology II, University of Bonn, Bonn, Germany. This article is distributed under the terms of the Creative Commons Attribution Noncommercial License (CC BY-NC 4.0) which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited. Abstract The neuromuscular disorder Duchenne muscular dystrophy is a multi-systemic disease that is caused by a primary abnormality in the X-chromosomal Dmd gene. Although progressive skeletal muscle wasting and cardio-respiratory complications are the most serious symptoms that are directly linked to the almost complete loss of the membrane cytoskeletal protein dystrophin, dystrophic patients also suffer from gastrointestinal dysfunction. In order to determine whether proteome-wide changes potentially occur in the gastrointestinal system due to dystrophin deficiency, total tissue extracts from the interface between the stomach wall and the pancreas of the mdx-4cv model of dystrophinopathy were analysed by mass spectrometry.