Thesis May Be Reproduced, Stored Or Transmitted in Any Form Or by Any Means, Without Permission of the Author
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mannosidases Are the Putative Catabolic Enzymes Which
THE CHARACTERIZATION OF A NOVEL HUMAN CORE-SPECIFIC LYSOSOMAL α1-6MANNOSIDASE INVOLVED IN N-GLYCAN CATABOLISM by CHAEHO PARK (Under the Direction of Kelley W. Moremen) ABSTRACT In humans and rodents lysosomal catabolism of Man3GlcNAc2 core N-glycan structures results from the concerted actions of exoglycosidases including the broad specificity lysosomal α- mannosidase (LysMan), a core-specific α1-6mannosidase, and β-mannosidase, as well as the core chitobiose cleavage by an di-N-acetylchitobiase. In ungulates and carnivora, both the chitobiase and the α1-6mannosidase enzyme activities are absent suggesting a co-regulation of the two enzymes. We describe here the cloning, expression, purification and characterization of the human core-specific α1-6mannosidase with similarity to members of the glycosylhydrolase family 38. The recombinant enzyme had a pH optimum of 4.0, was potently inhibited by swainsonine and 1,4-dideoxy1,4-imino-D-mannitol, and was stimulated by Co+2. NMR-based time course substrate specificity studies comparing the α1-6mannosidase with human LysMan revealed that the former enzyme selectively cleaved the α1-6mannose residue from Man3GlcNAc, but not Man3GlcNAc2 or other larger high mannose structures, indicating the requirement for chitobiase action prior to α1-6mannosidase cleavage. In contrast, LysMan cleaved all of the α-linked mannose residues from Man9-5GlcNAc2, Man3GlcNAc2, or Man3GlcNAc structures except the core α1-6mannose residue. Transcripts encoding the α1-6mannosidase were ubiquitously expressed in human tissues and expressed sequence tag searches in various mammalian species demonstrated a similar distribution in species-specific expression as the chitobiase. No expressed sequence tags were identified for bovine α1-6mannosidase despite the identification of two homologs in the bovine genome. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Molecular Cloning and Knockdown of Galactocerebrosidase in Zebrafish
Biochimica et Biophysica Acta 1842 (2014) 665–675 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbadis Molecular cloning and knockdown of galactocerebrosidase in zebrafish: New insights into the pathogenesis of Krabbe's disease Daniela Zizioli a,1, Michela Guarienti a,1,ChiaraTobiab, Giuseppina Gariano b, Giuseppe Borsani c, Roberto Bresciani a,RobertoRoncab, Edoardo Giacopuzzi c, Augusto Preti a, Germano Gaudenzi d, Mirella Belleri b, Emanuela Di Salle b, Gemma Fabrias e, Josefina Casas e,DomenicoRibattif,g, Eugenio Monti a,MarcoPrestab,⁎ a Unit of Biotechnology, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy b Unit of Oncology and Experimental Immunology, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy c Unit of Biology and Genetics, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy d Department of Biology, University of Milan, Milan, Italy e Research Unit on Bioactive Molecules (RUBAM), Department of Biomedicinal Chemistry, Institute for Advanced Chemistry of Catalonia (IQAC), Spanish Council for Scientific Research (CSIC), Barcelona, Spain f Department of Basic Biomedical Sciences, Unit of Human Anatomy and Histology, University of Bari, Bari, Italy g National Cancer Institute, Giovanni Paolo II, Bari, Italy article info abstract Article history: The lysosomal hydrolase galactocerebrosidase (GALC) catalyzes the removal of galactose from galactosylceramide Received 2 September 2013 and from other sphingolipids. GALC deficiency is responsible for globoid cell leukodystrophy (GLD), or Krabbe's Received in revised form 17 December 2013 disease, an early lethal inherited neurodegenerative disorder characterized by the accumulation of the neurotoxic Accepted 15 January 2014 metabolite psychosine in the central nervous system (CNS). -

Glycosphingolipids Are Modulators of Disease Pathogenesis in Amyotrophic Lateral Sclerosis
Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis James C. Dodgea,1, Christopher M. Treleavena, Joshua Pachecoa, Samantha Coopera,ChannaBaoa, Marissa Abrahama, Mandy Cromwella, S. Pablo Sardia, Wei-Lien Chuanga, Richard L. Sidmanb,1,2,SengH.Chenga, and Lamya S. Shihabuddina aRare Diseases Science, Genzyme, a Sanofi Company, Framingham, MA 01701; and bBeth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02215 Contributed by Richard L. Sidman, May 7, 2015 (sent for review January 12, 2015; reviewed by David V. Schaffer) Recent genetic evidence suggests that aberrant glycosphingolipid mediates the hydrolysis of Cer, are linked to forms of spinal metabolism plays an important role in several neuromuscular muscular atrophy that are not caused by the more frequent mu- diseases including hereditary spastic paraplegia, hereditary sen- tations in the survival motor neuron 1 gene (non-5q SMA) (15). sory neuropathy type 1, and non-5q spinal muscular atrophy. Here, Moreover, hereditary spastic paraplegia (HSP), a disease with cor- we investigated whether altered glycosphingolipid metabolism is ticospinal tract and in some cases, spinal cord MN degeneration, a modulator of disease course in amyotrophic lateral sclerosis (ALS). is attributed to mutations in GM2 synthase (16) and the non- Levels of ceramide, glucosylceramide, galactocerebroside, lactosyl- lysosomal glucosylceramidase, GBA2 (17). Last, patients with adult- ceramide, globotriaosylceramide, and the gangliosides GM3 and onset Tay-Sachs disease (GM2 gangliosidosis), a disease triggered GM1 were significantly elevated in spinal cords of ALS patients. by a deficiency in hexosaminidase (HEX), a lysosomal enzyme Moreover, enzyme activities (glucocerebrosidase-1, glucocerebrosi- that hydrolyzes GM2 to GM3, have been reported in some in- dase-2, hexosaminidase, galactosylceramidase, α-galactosidase, and stances to display a disease phenotype that closely mimics ALS β-galactosidase) mediating glycosphingolipid hydrolysis were also (18–20). -

GALC Gene Galactosylceramidase
GALC gene galactosylceramidase Normal Function The GALC gene provides instructions for making an enzyme called galactosylceramidase. Through a process called hydrolysis, this enzyme uses water molecules to break down certain fats called galactolipids, which are found primarily in the nervous system and kidneys. Within cells, galactosylceramidase is found in enzyme-filled sacs called lysosomes where it hydrolyzes specific galactolipids, including galactosylceramide and psychosine. Galactosylceramide is an important component of myelin, the protective covering around certain nerve cells that ensures the rapid transmission of nerve impulses. Its breakdown by galactosylceramidase is part of the normal turnover of myelin that occurs throughout life. Psychosine, which is toxic to cells, forms during the production of myelin and is quickly broken down by galactosylceramidase. Under normal conditions, tissues contain very little psychosine. Health Conditions Related to Genetic Changes Krabbe disease More than 200 GALC gene mutations that cause Krabbe disease have been identified. Krabbe disease is a brain disorder that usually begins in infancy (infantile Krabbe disease) and causes movement and eating problems, impaired development, and seizures. The most common mutation in affected individuals of European ancestry ( often called 30-kb del) deletes a large segment of the GALC gene. Other mutations insert additional DNA building blocks (nucleotides) into the GALC gene, delete a small number of nucleotides from the gene, or replace single nucleotides with incorrect nucleotides. These GALC gene mutations severely reduce or eliminate the activity of the galactosylceramidase enzyme. As a result, galactosylceramide and psychosine cannot be broken down. The accumulation of these galactolipids causes damage to myelin- forming cells, which impairs the formation of myelin and leads to the loss of myelin ( demyelination) in the nervous system. -

(12) United States Patent (10) Patent No.: US 9,689,046 B2 Mayall Et Al
USOO9689046B2 (12) United States Patent (10) Patent No.: US 9,689,046 B2 Mayall et al. (45) Date of Patent: Jun. 27, 2017 (54) SYSTEM AND METHODS FOR THE FOREIGN PATENT DOCUMENTS DETECTION OF MULTIPLE CHEMICAL WO O125472 A1 4/2001 COMPOUNDS WO O169245 A2 9, 2001 (71) Applicants: Robert Matthew Mayall, Calgary (CA); Emily Candice Hicks, Calgary OTHER PUBLICATIONS (CA); Margaret Mary-Flora Bebeselea, A. et al., “Electrochemical Degradation and Determina Renaud-Young, Calgary (CA); David tion of 4-Nitrophenol Using Multiple Pulsed Amperometry at Christopher Lloyd, Calgary (CA); Lisa Graphite Based Electrodes', Chem. Bull. “Politehnica” Univ. Kara Oberding, Calgary (CA); Iain (Timisoara), vol. 53(67), 1-2, 2008. Fraser Scotney George, Calgary (CA) Ben-Yoav. H. et al., “A whole cell electrochemical biosensor for water genotoxicity bio-detection”. Electrochimica Acta, 2009, 54(25), 6113-6118. (72) Inventors: Robert Matthew Mayall, Calgary Biran, I. et al., “On-line monitoring of gene expression'. Microbi (CA); Emily Candice Hicks, Calgary ology (Reading, England), 1999, 145 (Pt 8), 2129-2133. (CA); Margaret Mary-Flora Da Silva, P.S. et al., “Electrochemical Behavior of Hydroquinone Renaud-Young, Calgary (CA); David and Catechol at a Silsesquioxane-Modified Carbon Paste Elec trode'. J. Braz. Chem. Soc., vol. 24, No. 4, 695-699, 2013. Christopher Lloyd, Calgary (CA); Lisa Enache, T. A. & Oliveira-Brett, A. M., "Phenol and Para-Substituted Kara Oberding, Calgary (CA); Iain Phenols Electrochemical Oxidation Pathways”, Journal of Fraser Scotney George, Calgary (CA) Electroanalytical Chemistry, 2011, 1-35. Etesami, M. et al., “Electrooxidation of hydroquinone on simply prepared Au-Pt bimetallic nanoparticles'. Science China, Chem (73) Assignee: FREDSENSE TECHNOLOGIES istry, vol. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Chaperone Therapy for Krabbe Disease: Potential for Late-Onset GALC Mutations
Journal of Human Genetics (2015) 60, 539–545 & 2015 The Japan Society of Human Genetics All rights reserved 1434-5161/15 www.nature.com/jhg ORIGINAL ARTICLE Chaperone therapy for Krabbe disease: potential for late-onset GALC mutations Mohammad Arif Hossain1,7, Katsumi Higaki2, Seiji Saito3, Kazuki Ohno4,8, Hitoshi Sakuraba5, Eiji Nanba2, Yoshiyuki Suzuki6, Keiichi Ozono1 and Norio Sakai1 Krabbe disease is an autosomal recessive leukodystrophy caused by a deficiency of the galactocerebrosidase (GALC) enzyme. Hematopoietic stem cells transplantation is the only available treatment option for pre-symptomatic patients. We have previously reported the chaperone effect of N-octyl-4-epi-β-valienamine (NOEV) on mutant GM1 β-galactosidase proteins, and in a murine GM1-gangliosidosis model. In this study, we examined its chaperone effect on mutant GALC proteins. We found that NOEV strongly inhibited GALC activity in cell lysates of GALC-transfected COS1 cells. In vitro NOEV treatment stabilized GALC activity under heat denaturation conditions. We also examined the effect of NOEV on cultured COS1 cells expressing mutant GALC activity and human skin fibroblasts from Krabbe disease patients: NOEV significantly increased the enzyme activity of mutants of late-onset forms. Moreover, we confirmed that NOEV could enhance the maturation of GALC precursor to its mature active form. Model structural analysis showed NOEV binds to the active site of human GALC protein. These results, for the first time, provide clear evidence that NOEV is a chaperone with promising -

Altered Trafficking and Processing of GALC Mutants Correlates with Globoid Cell Leukodystrophy Severity
1858 • The Journal of Neuroscience, February 10, 2016 • 36(6):1858–1870 Neurobiology of Disease Altered Trafficking and Processing of GALC Mutants Correlates with Globoid Cell Leukodystrophy Severity Daesung Shin,1,2 M. Laura Feltri,1,2,3 and Lawrence Wrabetz1,2,3 1Hunter James Kelly Research Institute and Departments of 2Biochemistry and 3Neurology, Jacob School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, New York 14203 Globoid cell leukodystrophy (GLD, Krabbe disease) is due to autosomal recessive mutations in the lysosomal enzyme galactosylcerami- dase (GALC). Many GLD patients develop infantile-onset of progressive neurologic deterioration and death by 2 years of age, whereas others have a later-onset, milder disease. Cord blood transplant slows disease progression much more effectively when performed presymptomatically,highlightingtheimportanceofearlydiagnosis.CurrentdiagnosisisbasedonreducedGALCactivity,DNAsequence, and clinical examination. However, presymptomatic diagnosis is hampered by imperfect genotype-GALC activity-phenotype correla- tions. In addition, three polymorphisms in the GALC gene are variably associated with disease mutations and have unknown effects on GALC activity and disease outcome. Here, we study mutations that cause infantile or later-onset GLD, and show that GALC activity is significantly lower in infantile versus later-onset mutants when measured in the lysosomal fraction, but not in whole-cell lysates. In parallel, infantile-onset mutant GALCs showed reduced trafficking to lysosomes and processing than later-onset mutant GALCs. Finally, the cis-polymorphisms also affected trafficking to the lysosome and processing of GALC. These differences potentially explain why the activity of different mutations appears similar in whole-cell extracts from lymphocytes, and suggest that measure of GALC activity in lysosomes may better predict the onset and severity of disease for a given GLD genotype. -

The Unique Phenotype of Lipid-Laden Macrophages
International Journal of Molecular Sciences Review The Unique Phenotype of Lipid-Laden Macrophages Marco van Eijk * and Johannes M. F. G. Aerts * Leiden Institute of Chemistry, Leiden University, 2333 CC Leiden, The Netherlands * Correspondence: [email protected] (M.v.E.); [email protected] (J.M.F.G.A.) Abstract: Macrophages are key multi-talented cells of the innate immune system and are equipped with receptors involved in damage and pathogen recognition with connected immune response guiding signaling systems. In addition, macrophages have various systems that are involved in the uptake of extracellular and intracellular cargo. The lysosomes in macrophages play a central role in the digestion of all sorts of macromolecules and the entry of nutrients to the cytosol, and, thus, the regulation of endocytic processes and autophagy. Simplistically viewed, two macrophage phenotype extremes exist. On one end of the spectrum, the classically activated pro-inflammatory M1 cells are present, and, on the other end, alternatively activated anti-inflammatory M2 cells. A unique macrophage population arises when lipid accumulation occurs, either caused by flaws in the catabolic machinery, which is observed in lysosomal storage disorders, or as a result of an acquired condition, which is found in multiple sclerosis, obesity, and cardiovascular disease. The accompanying overload causes a unique metabolic activation phenotype, which is discussed here, and, consequently, a unifying phenotype is proposed. Keywords: adipose tissue; foam cell; Gaucher disease; GPNMB; macrophage; multiple sclerosis; obesity; TREM-2 Citation: van Eijk, M.; Aerts, J.M.F.G. The Unique Phenotype of Lipid-Laden Macrophages. -
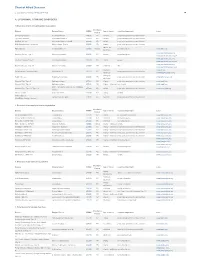
Pdf NTSAD Chart of Allied Diseases
Chart of Allied Diseases Last Updated: Monday, 19 May 2014 17:02 A. LYSOSOMAL STORAGE DISORDERS 1) Disorders of lipid and sphingolipid degradation Inheritance Disease Enzyme Defect OMIM# Age of Onset Cognitive Impairment Links Pattern GM1 Gangliosidosis b-Galactosidase-1 230500 AR variable progressive psychomotor deterioration Tay-Sachs Disease b-Hexosaminidase A 272800 AR variable progressive psychomotor deterioration Sandhoff Disease b-Hexosaminidases A and B 268800 AR variable progressive psychomotor deterioration GM2 Gangliodisosis, AB variant GM2 Activator Protein 272750 AR infancy progressive psychomotor deterioration adolesence - Fabry Disease 8-Galactosidase A 301500 X-linked normal intelligence www.fabry.org adulthood www.gaucherdisease.org, Gaucher Disease, Type 1 Glucocerebrosidase 230800 AR variable normal intelligence www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type II Glucocerebrosidase 230900 AR infancy severe www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type III Glucocerebrosidase 231000 AR childhood mild www.gaucherdisease.org.uk infancy to www.ulf.org, Metachromatic Leukodystrophy Arylsulfatase A 250100 AR progressive psychomotor deterioration adulthood www.MLDFoundation.org infancy to Krabbe Disease Galactosylceramidase 245200 AR progressive psychomotor deterioration www.huntershope.org adulthood Niemann-Pick, Type A Sphingomyelinase 257200 AR infancy progressive psychomotor deterioration www.nnpdf.org Niemann-Pick, Type B Sphingomyelinase 607616 AR infancy - childhood none -

Enzyme-Replacement Therapy in Lysosomal Storage
ENZYME.REPLACEMENT THERAPY IN LYSOSOMAL STORAGE DISEASES Julie Bielicki BSc Thesis submitted for the degree of Doctor of Philosophy 1n The University of Adelaide (F aculty of Medicine) Lysosomal Diseases Research Unit Department of Chemical PathologY 'Women's and Children's Hospital South Australia and Department of Paediatrics Faculty of Medicine University of Adelaide South Australia 2003 TABLE OF CONTENTS ABBREVIATIONS x THESIS ABSTRACT xv DECLARATION XViii ACKNO\MLEDGEMENTS xix Chanter 1: Introduction and review 1.1 Preface 1 1.2 Lysosomes 2 1.3 Lysosomal biogenesis J 1.4 Lysosomal enzymes 5 1.5 M6PR-mediated transport of lysosomal enzymes 6 1.6 M6PR-independent transport of lysosomal enzymes 8 1.7 Glycosaminoglycans and proteoglycans 8 1.7.1 Structure and function 8 1.7.2 Biosynthesis 11 1.7.3 Degradation t2 1.8 Glycoproteins T4 1.8.1 Structure and function I4 1.8.2 Biosynthesis 15 1.8.3 Degradation 15 1.9 Lysosomal storage diseases 18 1.10 The mucopolysaccharidoses t9 1.11 The oligosaccharidoses 2l 1.12 Human MPS VI 22 Ll2.l Historical 22 1'12.2 Genetics 22 1.12.3 Clinical 23 1.I2.4 Biochemistry 24 1.12.4.I Storage and PathologY 24 1.12.4.2 Enzymology 25 1.12.5 Diagnosis 25 1l 1.1,2.5.1 Urine analysis 26 1.12.5.2 45 activity 26 1.13 Bone synthesis and growth 27 1.13.1 Synthesis 27 1l3 .2 Elongation/growth 28 1.14 Human fucosidosis 29 1.14.1 Historical 29 |.14.2 Genetics 29 1.14.3 Clinical 30 1.14.4 Pathology 30 1.14.5 Diagnosis 31 1.15 Pathogenesis of LSD 31 1.16 Therapies for LSD JJ 1.16.1 Medical and surgical JJ 1.16.2 Organ and tissue transplantation 34 1.