View Receptors in Human Platelets by Surface Proteolysis with Elastase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Expression of Inflammation-Related Intercellular Adhesion Molecules in Cardiomyocytes in Vitro and Modulation by Pro-Inflammatory Agents
in vivo 30: 213-218 (2016) Expression of Inflammation-related Intercellular Adhesion Molecules in Cardiomyocytes In Vitro and Modulation by Pro-inflammatory Agents IBRAHIM EL-BATTRAWY1,2,3, EROL TÜLÜMEN2,3, SIEGFRIED LANG1,2,3, IBRAHIM AKIN2,3, MICHAEL BEHNES2,3, XIABO ZHOU1,2,3,4, MARTIN MAVANY2, PETER BUGERT5, KAREN BIEBACK5, MARTIN BORGGREFE2,3 and ELIF ELMAS2,3 1Division of Experimental Cardiology and 2First Department of Medicine, Medical Faculty Mannheim, University Heidelberg, Mannheim, Germany; 3German Center for Cardiovascular Research, Partner Site, Heidelberg-Mannheim, Mannheim, Germany; 4Institute of Cardiovascular Research, Sichuan Medical University, Luzhou, Sichuan, P.R. China; 5Institute for Transfusion Medicine and Immunology, Mannheim, Germany Abstract. Background: Cell-surface adhesion molecules not result in increased levels of CD31 (p>0.10). The pro- regulate multiple intercellular and intracellular processes and inflammatory agents LPS and thrombin had no effect on the play important roles in inflammation by facilitating leukocyte expression of MADCAM1 and F11R. Conclusion: endothelial transmigration. Whether cardiomyocytes express Inflammation-related cell-adhesion molecules CD31, surface-adhesion molecules related to inflammation and the MADCAM1 and F11R were shown to be expressed on the effect of pro-inflammatory mediators remain unknown. surface of human cardiomyocytes in an in vitro model. Materials and Methods: In the present study, the expression Incubation with LPS or thrombin resulted in increased of different cell-adhesion molecules (CD11a, CD11b, CD31, expression of CD31, however, it did not modify the expression CD62P, CD162, F11 receptor and mucosal vascular addressin of the cell adhesion molecules MADCAM1 and F11R. cell adhesion molecule 1 (MADCAM1)) and the effect of pro- inflammatory mediators were investigated in an in vitro model Coronary artery disease remains the leading cause of mortality of human cardiomyocytes. -

Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis
biomedicines Review Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis Thijs J. Sluiter 1,2 , Jaap D. van Buul 3 , Stephan Huveneers 4, Paul H. A. Quax 1,2 and Margreet R. de Vries 1,2,* 1 Department of Vascular Surgery, Leiden University Medical Center, 2333 ZA Leiden, The Netherlands; [email protected] (T.J.S.); [email protected] (P.H.A.Q.) 2 Einthoven Laboratory for Experimental Vascular Medicine, Leiden University Medical Center, 2333 ZA Leiden, The Netherlands 3 Sanquin Research and Landsteiner Laboratory, Leeuwenhoek Centre for Advanced Microscopy, Swammerdam Institute for Life Sciences, University of Amsterdam, 1066 CX Amsterdam, The Netherlands; [email protected] 4 Department of Medical Biochemistry, Amsterdam Cardiovascular Sciences, Amsterdam University Medical Center, Location AMC, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-(71)-526-5147 Abstract: The vascular endothelium is a highly specialized barrier that controls passage of fluids and migration of cells from the lumen into the vessel wall. Endothelial cells assist leukocytes to extravasate and despite the variety in the specific mechanisms utilized by different leukocytes to cross different vascular beds, there is a general principle of capture, rolling, slow rolling, arrest, crawling, and ultimately diapedesis via a paracellular or transcellular route. In atherosclerosis, the barrier function of the endothelium is impaired leading to uncontrolled leukocyte extravasation and Citation: Sluiter, T.J.; van Buul, J.D.; vascular leakage. This is also observed in the neovessels that grow into the atherosclerotic plaque Huveneers, S.; Quax, P.H.A.; de Vries, leading to intraplaque hemorrhage and plaque destabilization. -

Supplementary Materials and Tables a and B
SUPPLEMENTARY MATERIAL 1 Table A. Main characteristics of the subset of 23 AML patients studied by high-density arrays (subset A) WBC BM blasts MYST3- MLL Age/Gender WHO / FAB subtype Karyotype FLT3-ITD NPM status (x109/L) (%) CREBBP status 1 51 / F M4 NA 21 78 + - G A 2 28 / M M4 t(8;16)(p11;p13) 8 92 + - G G 3 53 / F M4 t(8;16)(p11;p13) 27 96 + NA G NA 4 24 / M PML-RARα / M3 t(15;17) 5 90 - - G G 5 52 / M PML-RARα / M3 t(15;17) 1.5 75 - - G G 6 31 / F PML-RARα / M3 t(15;17) 3.2 89 - - G G 7 23 / M RUNX1-RUNX1T1 / M2 t(8;21) 38 34 - + ND G 8 52 / M RUNX1-RUNX1T1 / M2 t(8;21) 8 68 - - ND G 9 40 / M RUNX1-RUNX1T1 / M2 t(8;21) 5.1 54 - - ND G 10 63 / M CBFβ-MYH11 / M4 inv(16) 297 80 - - ND G 11 63 / M CBFβ-MYH11 / M4 inv(16) 7 74 - - ND G 12 59 / M CBFβ-MYH11 / M0 t(16;16) 108 94 - - ND G 13 41 / F MLLT3-MLL / M5 t(9;11) 51 90 - + G R 14 38 / F M5 46, XX 36 79 - + G G 15 76 / M M4 46 XY, der(10) 21 90 - - G NA 16 59 / M M4 NA 29 59 - - M G 17 26 / M M5 46, XY 295 92 - + G G 18 62 / F M5 NA 67 88 - + M A 19 47 / F M5 del(11q23) 17 78 - + M G 20 50 / F M5 46, XX 61 59 - + M G 21 28 / F M5 46, XX 132 90 - + G G 22 30 / F AML-MD / M5 46, XX 6 79 - + M G 23 64 / M AML-MD / M1 46, XY 17 83 - + M G WBC: white blood cell. -

PAR1 Agonists Stimulate APC-Like Endothelial Cytoprotection and Confer Resistance to Thromboinflammatory Injury
PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury Karen De Ceunyncka,1, Christian G. Petersa,1, Abhishek Jainb,2, Sarah J. Higginsc,d, Omozuanvbo Aisikua, Jennifer L. Fitch-Tewfika, Sharjeel A. Chaudhrya, Chris Dockendorffe, Samir M. Parikhc,d, Donald E. Ingberb,f,g,h, and Robert Flaumenhafta,3 aDivision of Hemostasis and Thrombosis, Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02115; bWyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115; cDivision of Nephrology, Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02115; dCenter for Vascular Biology Research, Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02115; eDepartment of Chemistry, Marquette University, Milwaukee, WI 53201; fVascular Biology Program, Boston Children’s Hospital and Harvard Medical School, Boston, MA 02115; gDepartment of Surgery, Boston Children’s Hospital and Harvard Medical School, Boston, MA 02115; and hHarvard John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138 Edited by Barry S. Coller, The Rockefeller University, New York, NY, and approved December 18, 2017 (received for review November 1, 2017) Stimulation of protease-activated receptor 1 (PAR1) on endothelium results in the generation of a unique tethered ligand that stimu- by activated protein C (APC) is protective in several animal models lates antiinflammatory, antiapoptotic, and barrier function for- of disease, and APC has been used clinically in severe sepsis and tifying pathways in endothelium (10, 18–23). Antithrombotic wound healing. Clinical use of APC, however, is limited by its activity may also be a component of this response. -

Gene Expression and Functionality in Head and Neck Cancer
ONCOLOGY REPORTS 35: 2431-2440, 2016 Spheroid-based 3-dimensional culture models: Gene expression and functionality in head and neck cancer MARIANNE SCHMIDT1, CLAUS-JUERGEN SCHOLZ2, CHRISTINE POLEDNIK1 and JEANETTE ROLLER1 1Department of Otorhinolaryngology, University of Würzburg, D-97080 Würzburg; 2Interdisciplinary Center for Clinical Research, Microarray Core Unit, University of Würzburg, D-97078 Würzburg, Germany Received October 26, 2015; Accepted December 5, 2015 DOI: 10.3892/or.2016.4581 Abstract. In the present study a panel of 12 head and neck Introduction cancer (HNSCC) cell lines were tested for spheroid forma- tion. Since the size and morphology of spheroids is dependent According to an analysis of over 3,000 cases of primary head on both cell adhesion and proliferation in the 3-dimensional and neck tumours in Germany in 2009, the patient benefit has (3D) context, morphology of HNSCC spheroids was related to not significantly improved from 1995 to 2006, despite new expression of E-cadherin and the proliferation marker Ki67. In treatment strategies. Particularly, the 5-year overall survival HNSCC cell lines the formation of tight regular spheroids was rate for carcinomas of hypopharyngeal origin is very low with dependent on distinct E-cadherin expression levels in mono- 27.2% (1). Hypopharyngeal cancer is often detected in later layer cultures, usually resulting in upregulation following stages, since early signs and symptoms rarely occur. Late aggregation into 3D structures. Cell lines expressing only low stages frequently involve multiple node affection, leading levels of E-cadherin in monolayers produced only loose cell to poor prognosis (http://emedicine.medscape.com/article clusters, frequently decreasing E-cadherin expression further /1375268). -

Differences in Gene Expression Profiles and Carcinogenesis Pathways Involved in Cisplatin Resistance of Four Types of Cancer
596 ONCOLOGY REPORTS 30: 596-614, 2013 Differences in gene expression profiles and carcinogenesis pathways involved in cisplatin resistance of four types of cancer YONG YANG1,2, HUI LI1,2, SHENGCAI HOU1,2, BIN HU1,2, JIE LIU1,3 and JUN WANG1,3 1Beijing Key Laboratory of Respiratory and Pulmonary Circulation, Capital Medical University, Beijing 100069; 2Department of Thoracic Surgery, Beijing Chao-Yang Hospital, Capital Medical University, Beijing 100020; 3Department of Physiology, Capital Medical University, Beijing 100069, P.R. China Received December 23, 2012; Accepted March 4, 2013 DOI: 10.3892/or.2013.2514 Abstract. Cisplatin-based chemotherapy is the standard Introduction therapy used for the treatment of several types of cancer. However, its efficacy is largely limited by the acquired drug Cisplatin is primarily effective through DNA damage and is resistance. To date, little is known about the RNA expression widely used for the treatment of several types of cancer, such changes in cisplatin-resistant cancers. Identification of the as testicular, lung and ovarian cancer. However, the ability RNAs related to cisplatin resistance may provide specific of cancer cells to become resistant to cisplatin remains a insight into cancer therapy. In the present study, expression significant impediment to successful chemotherapy. Although profiling of 7 cancer cell lines was performed using oligo- previous studies have identified numerous mechanisms in nucleotide microarray analysis data obtained from the GEO cisplatin resistance, it remains a major problem that severely database. Bioinformatic analyses such as the Gene Ontology limits the usefulness of this chemotherapeutic agent. Therefore, (GO) and KEGG pathway were used to identify genes and it is crucial to examine more elaborate mechanisms of cisplatin pathways specifically associated with cisplatin resistance. -

Mouse CD Marker Chart Bdbiosciences.Com/Cdmarkers
BD Mouse CD Marker Chart bdbiosciences.com/cdmarkers 23-12400-01 CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD1d CD1.1, CD1.2, Ly-38 Lipid, Glycolipid Ag + + + + + + + + CD104 Integrin b4 Laminin, Plectin + DNAX accessory molecule 1 (DNAM-1), Platelet and T cell CD226 activation antigen 1 (PTA-1), T lineage-specific activation antigen 1 CD112, CD155, LFA-1 + + + + + – + – – CD2 LFA-2, Ly-37, Ly37 CD48, CD58, CD59, CD15 + + + + + CD105 Endoglin TGF-b + + antigen (TLiSA1) Mucin 1 (MUC1, MUC-1), DF3 antigen, H23 antigen, PUM, PEM, CD227 CD54, CD169, Selectins; Grb2, β-Catenin, GSK-3β CD3g CD3g, CD3 g chain, T3g TCR complex + CD106 VCAM-1 VLA-4 + + EMA, Tumor-associated mucin, Episialin + + + + + + Melanotransferrin (MT, MTF1), p97 Melanoma antigen CD3d CD3d, CD3 d chain, T3d TCR complex + CD107a LAMP-1 Collagen, Laminin, Fibronectin + + + CD228 Iron, Plasminogen, pro-UPA (p97, MAP97), Mfi2, gp95 + + CD3e CD3e, CD3 e chain, CD3, T3e TCR complex + + CD107b LAMP-2, LGP-96, LAMP-B + + Lymphocyte antigen 9 (Ly9), -

Differential Co-Expression Analysis of Obesity-Associated Networks in Human Subcutaneous Adipose Tissue
Europe PMC Funders Group Author Manuscript Int J Obes (Lond). Author manuscript; available in PMC 2012 July 01. Published in final edited form as: Int J Obes (Lond). 2012 January ; 36(1): 137–147. doi:10.1038/ijo.2011.22. Europe PMC Funders Author Manuscripts Differential co-expression analysis of obesity-associated networks in human subcutaneous adipose tissue A.J. Walley1,*, P. Jacobson2,*, M. Falchi1,*, L. Bottolo1,3, J.C. Andersson1,2, E. Petretto3,4, A. Bonnefond5, E. Vaillant5, C. Lecoeur5, V. Vatin5, M. Jernas2, D. Balding1,6, M. Petteni1, Y.S. Park1, T. Aitman4, S. Richardson3, L. Sjostrom2, L. M. S. Carlsson2,*, and P. Froguel1,5,*,† 1 Department of Genomics of Common Disease, School of Public Health, Imperial College London, Hammersmith Hospital, Du Cane Road, London, W12 0NN, UK 2 Department of Molecular and Clinical Medicine, The Sahlgrenska Academy, Gothenburg University, SE-413 07 Gothenburg, Sweden 3 Department of Epidemiology and Biostatistics, School of Public Health, Imperial College London, St Marys Hospital, 161 Norfolk Place, London, UK 4 MRC Clinical Sciences Centre, Division of Clinical Sciences, Imperial College London, Commonwealth Building, Hammersmith Hospital, Du Cane Road, London, W12 0NN, UK 5CNRS 8090-Institute of Biology, Pasteur Institute, Lille, France. 6Institute of Genetics, University College London, Kathleen Lonsdale Building, 5 Gower Place, London, WC1 E6B, UK Abstract Europe PMC Funders Author Manuscripts Objective—To use a unique obesity-discordant sib-pair study design to combine differential expression analysis, expression quantitative trait loci (eQTLs) mapping, and a co-expression regulatory network approach in subcutaneous human adipose tissue to identify genes relevant to the obese state. -
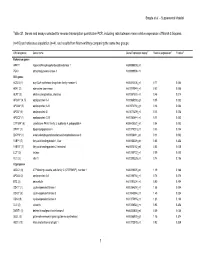
Table S1. Genes and Assays Selected for Reverse Transcription Quantitative PCR, Including Ratio Between Mean Relative Expression of Marsh 3 Biopsies
Bragde et al. – Supplemental Material Table S1. Genes and assays selected for reverse transcription quantitative PCR, including ratio between mean relative expression of Marsh 3 biopsies (n=10) and reference population (n=4), and results from Mann-whitney comparing the same two groups. Official symbol Gene name Gene Expression assay** Relative expression‡ P-value§ Reference genes HPRT1 hypoxanthine phosphoribosyltransferase 1 Hs99999909_m1 PGK1 phosphoglycerate kinase 1 Hs99999906_m1 Villi genes ACSL5 (1) acyl-CoA synthetase long-chain family member 5 Hs00212106_m1 0.77 0.054 ADA* (2) adenosine deaminase Hs01110949_m1 0.52 0.008 ALPI* (3) alkaline phosphatase, intestinal Hs00357578_m1 0.46 0.014 APOA1* (4, 5) apolipoprotein A-I Hs00985000_g1 0.05 0.002 APOA4* (5) apolipoprotein A-IV Hs01573751_g1 0.16 0.002 APOB* (5) apolipoprotein B Hs01071209_m1 0.13 0.002 APOC3* (1) apolipoprotein C-III Hs00163644_m1 0.11 0.002 CYP3A4* (6) cytochrome P450, family 3, subfamily A, polypeptide 4 Hs00430021_m1 0.04 0.002 DPP4* (1) dipeptidyl-peptidase 4 Hs00175210_m1 0.33 0.004 ENPP3* (1) ectonucleotide pyrophosphatase/phosphodiesterase 3 Hs01038411_m1 0.11 0.002 FABP1 (7) fatty acid binding protein 1, liver Hs00155026_m1 0.85 0.454 FABP2* (7) fatty acid binding protein 2, intestinal Hs01573162_m1 0.53 0.008 LCT* (2) lactase Hs00158722_m1 0.09 0.002 VIL1 (2) villin 1 Hs00200229_m1 0.74 0.106 Crypt genes ABCC1 (8) ATP-binding cassette, sub-family C (CFTR/MRP), member 1 Hs00219905_m1 1.19 0.188 APOA2 (2) apolipoprotein A-II Hs00155788_m1 0.78 0.076 BTC (2) betacellulin Hs01101204_m1 0.90 0.454 CDK1* (1) cyclin-dependent kinase 1 Hs00364293_m1 1.58 0.004 CDK2* (9) cyclin-dependent kinase 2 Hs01548894_m1 1.45 0.024 CDK4 (9) cyclin-dependent kinase 4 Hs00175935_m1 1.23 0.188 CLU (2) clusterin Hs00156548_m1 0.93 0.454 DMBT1 (1) deleted in malignant brain tumors 1 Hs00244838_m1 0.69 0.054 GLUL (2) glutamate-ammonia ligase (glutamine synthetase) Hs00365928_g1 1.15 0.374 HES1 (10) Hairy and enhancer of split 1 Hs00172878_m1 0.92 0.839 1 Bragde et al.