Virus Discovery in All Three Major Lineages of Terrestrial Arthropods Highlights the Diversity of Single-Stranded DNA Viruses Associated with Invertebrates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Study Guide Entomology & Nematology Department
STUDY GUIDE ENTOMOLOGY & NEMATOLOGY DEPARTMENT DPM COMPREHENSIVE EXAMINATIONS The Entomology & Nematology Comprehensive Examinations consist of 3 sections: pest identification (30%), pest biology and management (40%), and core concepts and synthesis (30%). These examinations are limited to information about invertebrate animal pests, principally insects and nematodes, but also plant feeding mites and terrestrial molluscs. A. Pest identification Students will be presented with insects, mites, molluscs, and nematodes that they must identify. Some may be recognizable by sight, but others may require keys for identification. Students will be provided with identification aids (keys), where necessary, and be expected to use them to identify the subjects accurately. The unknowns will be selected from the list of important insect, mite, mollusc, and nematode pests (Table 1) though we will emphasize those with a single or double asterisk [* or **]), as these normally are the more important pests. Included in this list are some that pose a threat but are not currently found in Florida. B. Pest biology and management Students will answer 8-10 questions on insect, mite, mollusc, and nematode pest biology (sampling, distribution, life cycle, damage) and management. The animals for which students are responsible to know biology and management are listed in Table 1 (preceded by double asterisk [**]). C. Core Concepts and Synthesis Section: Students will answer 3 or 4 questions that cover core areas of Entomology/Nematology and demonstrate knowledge of core areas, but also analysis and problem solving. Suggested reference/reading material is listed in Table 2. You might want to read through these in preparation for the Comprehensive Examinations. -

Pdf Available
Virology 554 (2021) 89–96 Contents lists available at ScienceDirect Virology journal homepage: www.elsevier.com/locate/virology Diverse cressdnaviruses and an anellovirus identifiedin the fecal samples of yellow-bellied marmots Anthony Khalifeh a, Daniel T. Blumstein b,**, Rafaela S. Fontenele a, Kara Schmidlin a, C´ecile Richet a, Simona Kraberger a, Arvind Varsani a,c,* a The Biodesign Center for Fundamental and Applied Microbiomics, School of Life Sciences, Center for Evolution and Medicine, Arizona State University, Tempe, AZ, 85287, USA b Department of Ecology & Evolutionary Biology, Institute of the Environment & Sustainability, University of California Los Angeles, Los Angeles, CA, 90095, USA c Structural Biology Research Unit, Department of Clinical Laboratory Sciences, University of Cape Town, 7925, Cape Town, South Africa ARTICLE INFO ABSTRACT Keywords: Over that last decade, coupling multiple strand displacement approaches with high throughput sequencing have Marmota flaviventer resulted in the identification of genomes of diverse groups of small circular DNA viruses. Using a similar Anelloviridae approach but with recovery of complete genomes by PCR, we identified a diverse group of single-stranded vi Genomoviridae ruses in yellow-bellied marmot (Marmota flaviventer) fecal samples. From 13 fecal samples we identified viruses Cressdnaviricota in the family Genomoviridae (n = 7) and Anelloviridae (n = 1), and several others that ware part of the larger Cressdnaviricota phylum but not within established families (n = 19). There were also circular DNA molecules identified (n = 4) that appear to encode one viral-like gene and have genomes of <1545 nts. This study gives a snapshot of viruses associated with marmots based on fecal sampling. -

Viral Diversity in Oral Cavity from Sapajus Nigritus by Metagenomic Analyses
Brazilian Journal of Microbiology (2020) 51:1941–1951 https://doi.org/10.1007/s42770-020-00350-w ENVIRONMENTAL MICROBIOLOGY - RESEARCH PAPER Viral diversity in oral cavity from Sapajus nigritus by metagenomic analyses Raissa Nunes dos Santos1,2 & Fabricio Souza Campos2,3 & Fernando Finoketti1,2 & Anne Caroline dos Santos1,2 & Aline Alves Scarpellini Campos1,2,3 & Paulo Guilherme Carniel Wagner2,4 & Paulo Michel Roehe 1,2 & Helena Beatriz de Carvalho Ruthner Batista2,5 & Ana Claudia Franco1,2 Received: 20 January 2020 /Accepted: 25 July 2020 / Published online: 11 August 2020 # Sociedade Brasileira de Microbiologia 2020 Abstract Sapajus nigritus are non-human primates which are widespread in South America. They are omnivores and live in troops of up to 40 individuals. The oral cavity is one of the main entry routes for microorganisms, including viruses. Our study proposed the identification of viral sequences from oral swabs collected in a group of capuchin monkeys (n = 5) living in a public park in a fragment of Mata Atlantica in South Brazil. Samples were submitted to nucleic acid extraction and enrichment, which was followed by the construction of libraries. After high-throughput sequencing and contig assembly, we used a pipeline to identify 11 viral families, which are Herpesviridae, Parvoviridae, Papillomaviridae, Polyomaviridae, Caulimoviridae, Iridoviridae, Astroviridae, Poxviridae,andBaculoviridae, in addition to two complete viral genomes of Anelloviridae and Genomoviridae. Some of these viruses were closely related to known viruses, while other fragments are more distantly related, with 50% of identity or less to the currently available virus sequences in databases. In addition to host-related viruses, insect and small vertebrate-related viruses were also found, as well as plant-related viruses, bringing insights about their diet. -

Distribution and Population Dynamics of the Asian Cockroach
DISTRIBUTION AND POPULATION DYNAMICS OF THE ASIAN COCKROACH (BLATTELLA ASAHINIA MIZUKUBO) IN SOUTHERN ALABAMA AND GEORGIA Except where reference is made to the work of others, the work described in this thesis is my own or was done in collaboration with my advisory committee. This thesis does not include proprietary or classified information. ___________________________________ Edward Todd Snoddy Certificate of Approval: ___________________________ ___________________________ Micky D. Eubanks Arthur G. Appel, Chair Associate Professor Professor Entomology and Plant Pathology Entomology and Plant Pathology ___________________________ ___________________________ Xing Ping Hu George T. Flowers Associate Professor Interim Dean Entomology and Plant Pathology Graduate School DISTRIBUTION AND POPULATION DYNAMICS OF THE ASIAN COCKROACH (BLATTELLA ASAHINIA MIZUKUBO) IN SOUTHERN ALABAMA AND GEORGIA Edward Todd Snoddy A Thesis Submitted to the Graduate Faculty of Auburn University in Partial Fulfillment of the Requirements for the Degree of Master of Science Auburn, Alabama May 10, 2007 DISTRIBUTION AND POPULATION DYNAMICS OF THE ASIAN COCKROACH (BLATTELLA ASAHINIA MIZUKUBO) IN SOUTHERN ALABAMA AND GEORGIA Edward Todd Snoddy Permission is granted to Auburn University to make copies of this thesis at its discretion, upon request of individuals or institutions and at their expense. The author reserves all publication rights. _______________________ Signature of Author _______________________ Date of Graduation iii VITA Edward Todd Snoddy was born in Auburn, Alabama on February 28, 1964 to Dr. Edward Lewis Snoddy and Lucy Mae Snoddy. He graduated Sheffield High School, Sheffield, Alabama in 1981. He attended Alexander Junior College from 1981 to 1983 at which time he transferred to Auburn University. He married Tracy Smith of Uchee, Alabama in 1984. -

Ephydra Hians) Say at Mono Lake, California (USA) in Relation to Physical Habitat
Hydrobiologia 197: 193-205, 1990. F. A. Comln and T. G. Northcote (eds), Saline Lakes. 193 © 1990 Kluwer Academic Publishers. Printed in Belgium. Distribution and abundance of the alkali fly (Ephydra hians) Say at Mono Lake, California (USA) in relation to physical habitat David B. Herbst Sierra Nevada Aquatic Research Laboratory, University of California, Star Route 1, Box 198, Mammoth Lakes, CA 93546, USA Key words: Ephydra, life cycle, development, distribution, Mono Lake, substrate Abstract The distribution and abundance of larval, pupal, and adult stages of the alkali fly Ephydra hians Say were examined in relation to location, benthic substrate type, and shoreline features at Mono Lake. Generation time was calculated as a degree-day model for development time at different temperatures, and compared to the thermal environment of the lake at different depths. Larvae and pupae have a contagious distribution and occur in greatest abundance in benthic habitats containing tufa (a porous limestone deposit), and in least abundance on sand or sand/mud substrates. Numbers increase with increasing area of tufa present in a sample, but not on other rocky substrates (alluvial gravel/cobble or cemented sand). Standing stock densities are greatest at locations around the lake containing a mixture of tufa deposits, detrital mud sediments, and submerged vegetation. Shoreline adult abundance is also greatest in areas adjacent to tufa. The shore fly (ephydrid) community varies in composition among different shoreline habitats and shows a zonation with distance from shore. The duration of pupation (from pupa formation to adult eclosion) becomes shorter as temperature increases. The temperature dependence of pupa development time is not linear and results in prolonged time requirements to complete development at temperatures below 20 ° C. -

Multiple Origins of Prokaryotic and Eukaryotic Single-Stranded DNA Viruses from Bacterial and Archaeal Plasmids
ARTICLE https://doi.org/10.1038/s41467-019-11433-0 OPEN Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids Darius Kazlauskas 1, Arvind Varsani 2,3, Eugene V. Koonin 4 & Mart Krupovic 5 Single-stranded (ss) DNA viruses are a major component of the earth virome. In particular, the circular, Rep-encoding ssDNA (CRESS-DNA) viruses show high diversity and abundance 1234567890():,; in various habitats. By combining sequence similarity network and phylogenetic analyses of the replication proteins (Rep) belonging to the HUH endonuclease superfamily, we show that the replication machinery of the CRESS-DNA viruses evolved, on three independent occa- sions, from the Reps of bacterial rolling circle-replicating plasmids. The CRESS-DNA viruses emerged via recombination between such plasmids and cDNA copies of capsid genes of eukaryotic positive-sense RNA viruses. Similarly, the rep genes of prokaryotic DNA viruses appear to have evolved from HUH endonuclease genes of various bacterial and archaeal plasmids. Our findings also suggest that eukaryotic polyomaviruses and papillomaviruses with dsDNA genomes have evolved via parvoviruses from CRESS-DNA viruses. Collectively, our results shed light on the complex evolutionary history of a major class of viruses revealing its polyphyletic origins. 1 Institute of Biotechnology, Life Sciences Center, Vilnius University, Saulėtekio av. 7, Vilnius 10257, Lithuania. 2 The Biodesign Center for Fundamental and Applied Microbiomics, School of Life Sciences, Center for Evolution and Medicine, Arizona State University, Tempe, AZ 85287, USA. 3 Structural Biology Research Unit, Department of Integrative Biomedical Sciences, University of Cape Town, Rondebosch, 7700 Cape Town, South Africa. -

Primordial Capsid and Spooled Ssdna Genome Structures Penetrate
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.14.435335; this version posted March 14, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Primordial capsid and spooled ssDNA genome structures penetrate 2 ancestral events of eukaryotic viruses 3 4 Anna Munke1*#, Kei Kimura2, Yuji Tomaru3, Han Wang1, Kazuhiro Yoshida4, Seiya Mito5, Yuki 5 Hongo6, and Kenta Okamoto1* 6 1. The Laboratory of Molecular Biophysics, Department of Cell and Molecular Biology, Uppsala 7 University, Uppsala, Sweden 8 2. Department of Biological Resource Science, Faculty of Agriculture, Saga University, Saga, Japan 9 3. Fisheries Technology Institute, Japan Fisheries Research and Education Agency, Hatsukaichi, 10 Hiroshima, Japan 11 4. Graduate School of Agriculture, Saga University, Saga, Japan 12 5. Department of Biological Resource Science, Faculty of Agriculture, Saga University, Saga, Japan 13 6. Bioinformatics and Biosciences Division, Fisheries Resources Institute, Japan Fisheries Research 14 and Education Agency, Fukuura, Kanazawa, Yokohama, Kanagawa, Japan 15 16 17 *Corresponding authors 18 Corresponding author 1: [email protected] 19 Corresponding author 2: [email protected] 20 21 22 #Present address: Center for Free Electron Laser Science, Deutsches Elektronen-Synchrotron DESY, 23 Hamburg 22607, Germany 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.14.435335; this version posted March 14, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 24 Abstract 25 Marine algae viruses are important for controlling microorganism communities in the marine 26 ecosystem, and played a fundamental role during the early events of viral evolution. -

Beet Curly Top Virus Strains Associated with Sugar Beet in Idaho, Oregon, and a Western U.S
Plant Disease • 2017 • 101:1373-1382 • http://dx.doi.org/10.1094/PDIS-03-17-0381-RE Beet curly top virus Strains Associated with Sugar Beet in Idaho, Oregon, and a Western U.S. Collection Carl A. Strausbaugh and Imad A. Eujayl, United States Department of Agriculture–Agricultural Research Service (USDA-ARS) Northwest Irrigation and Soils Research Laboratory, Kimberly, ID 83341; and William M. Wintermantel, USDA-ARS, Salinas, CA 93905 Abstract Curly top of sugar beet is a serious, yield-limiting disease in semiarid pro- Logan) strains and primers that amplified a group of Worland (Wor)- duction areas caused by Beet curly top virus (BCTV) and transmitted like strains. The BCTV strain distribution averaged 2% Svr, 30% CA/ by the beet leafhopper. One of the primary means of control for BCTV Logan, and 87% Wor-like (16% had mixed infections), which differed in sugar beet is host resistance but effectiveness of resistance can vary from the previously published 2006-to-2007 collection (87% Svr, 7% among BCTV strains. Strain prevalence among BCTV populations CA/Logan, and 60% Wor-like; 59% mixed infections) based on a contin- was last investigated in Idaho and Oregon during a 2006-to-2007 collec- gency test (P < 0.0001). Whole-genome sequencing (GenBank acces- tion but changes in disease severity suggested a need for reevaluation. sions KT276895 to KT276920 and KX867015 to KX867057) with Therefore, 406 leaf samples symptomatic for curly top were collected overlapping primers found that the Wor-like strains included Wor, Colo- from sugar beet plants in commercial sugar beet fields in Idaho and rado and a previously undescribed strain designated Kimberly1. -

Novel Circular DNA Viruses in Stool Samples of Wild-Living Chimpanzees
Journal of General Virology (2010), 91, 74–86 DOI 10.1099/vir.0.015446-0 Novel circular DNA viruses in stool samples of wild-living chimpanzees Olga Blinkova,1 Joseph Victoria,1 Yingying Li,2 Brandon F. Keele,2 Crickette Sanz,33 Jean-Bosco N. Ndjango,4 Martine Peeters,5 Dominic Travis,6 Elizabeth V. Lonsdorf,7 Michael L. Wilson,8,9 Anne E. Pusey,9 Beatrice H. Hahn2 and Eric L. Delwart1 Correspondence 1Blood Systems Research Institute, San Francisco and the Department of Laboratory Medicine, Eric L. Delwart University of California, San Francisco, CA, USA [email protected] 2Departments of Medicine and Microbiology, University of Alabama at Birmingham, Birmingham, AL, USA 3Max-Planck Institute for Evolutionary Anthropology, Leipzig, Germany 4Department of Ecology and Management of Plant and Animal Ressources, Faculty of Sciences, University of Kisangani, Democratic Republic of the Congo 5UMR145, Institut de Recherche pour le De´velopement and University of Montpellier 1, Montpellier, France 6Department of Conservation and Science, Lincoln Park Zoo, Chicago, IL 60614, USA 7The Lester E. Fisher Center for the Study and Conservation of Apes, Lincoln Park Zoo, Chicago, IL 60614, USA 8Department of Anthropology, University of Minnesota, Minneapolis, MN 55455, USA 9Jane Goodall Institute’s Center for Primate Studies, Department of Ecology, Evolution and Behavior, University of Minnesota, St Paul, MN 55108, USA Viral particles in stool samples from wild-living chimpanzees were analysed using random PCR amplification and sequencing. Sequences encoding proteins distantly related to the replicase protein of single-stranded circular DNA viruses were identified. Inverse PCR was used to amplify and sequence multiple small circular DNA viral genomes. -
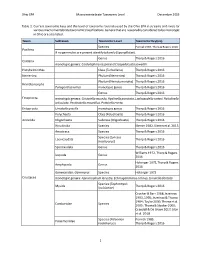
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 1 Table 1. Current Taxonomic Keys and the Level of Taxonomy Routinely U
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Table 1. Current taxonomic keys and the level of taxonomy routinely used by the Ohio EPA in streams and rivers for various macroinvertebrate taxonomic classifications. Genera that are reasonably considered to be monotypic in Ohio are also listed. Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Species Pennak 1989, Thorp & Rogers 2016 Porifera If no gemmules are present identify to family (Spongillidae). Genus Thorp & Rogers 2016 Cnidaria monotypic genera: Cordylophora caspia and Craspedacusta sowerbii Platyhelminthes Class (Turbellaria) Thorp & Rogers 2016 Nemertea Phylum (Nemertea) Thorp & Rogers 2016 Phylum (Nematomorpha) Thorp & Rogers 2016 Nematomorpha Paragordius varius monotypic genus Thorp & Rogers 2016 Genus Thorp & Rogers 2016 Ectoprocta monotypic genera: Cristatella mucedo, Hyalinella punctata, Lophopodella carteri, Paludicella articulata, Pectinatella magnifica, Pottsiella erecta Entoprocta Urnatella gracilis monotypic genus Thorp & Rogers 2016 Polychaeta Class (Polychaeta) Thorp & Rogers 2016 Annelida Oligochaeta Subclass (Oligochaeta) Thorp & Rogers 2016 Hirudinida Species Klemm 1982, Klemm et al. 2015 Anostraca Species Thorp & Rogers 2016 Species (Lynceus Laevicaudata Thorp & Rogers 2016 brachyurus) Spinicaudata Genus Thorp & Rogers 2016 Williams 1972, Thorp & Rogers Isopoda Genus 2016 Holsinger 1972, Thorp & Rogers Amphipoda Genus 2016 Gammaridae: Gammarus Species Holsinger 1972 Crustacea monotypic genera: Apocorophium lacustre, Echinogammarus ischnus, Synurella dentata Species (Taphromysis Mysida Thorp & Rogers 2016 louisianae) Crocker & Barr 1968; Jezerinac 1993, 1995; Jezerinac & Thoma 1984; Taylor 2000; Thoma et al. Cambaridae Species 2005; Thoma & Stocker 2009; Crandall & De Grave 2017; Glon et al. 2018 Species (Palaemon Pennak 1989, Palaemonidae kadiakensis) Thorp & Rogers 2016 1 Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Informal grouping of the Arachnida Hydrachnidia Smith 2001 water mites Genus Morse et al. -

Ocorrência Natural De Espécies Virais E Avaliação De Potenciais Hospedeiras Experimentais De Begomovírus, Potyvírus E Tospovírus Em Plantas Arbóreas E Berinjela
UNIVERSIDADE DE BRASÍLIA INSTITUTO DE CIÊNCIAS BIOLÓGICAS DEPARTAMENTO DE FITOPATOLOGIA PROGRAMA DE PÓS-GRADUAÇÃO EM FITOPATOLOGIA Ocorrência natural de espécies virais e avaliação de potenciais hospedeiras experimentais de begomovírus, potyvírus e tospovírus em plantas arbóreas e berinjela (Solanum melongena) JOSIANE GOULART BATISTA Brasília - DF 2014 JOSIANE GOULART BATISTA Ocorrência natural de espécies virais e avaliação de potenciais hospedeiras experimentais de begomovírus, potyvírus e tospovírus em plantas arbóreas e berinjela (Solanum melongena) Dissertação apresentada à Universidade de Brasília como requisito parcial para a obtenção do título de Mestre em Fitopatologia pelo Programa de Pós Graduação em Fitopatologia. Orientadora Dra. Rita de Cássia Pereira Carvalho BRASÍLIA DISTRITO FEDERAL – BRASIL 2014 FICHA CATALOGRÁFICA Batista, G.J. Ocorrência natural de espécies virais e avaliação de potenciais hospedeiras experimentais de begomovírus, potyvírus e tospovírus em plantas arbóreas e berinjela (Solanum melongena). Josiane Goulart Batista. Brasília, 2014 Número de páginas p.: 183 Dissertação de mestrado - Programa de Pós-Graduação em Fitopatologia, Universidade de Brasília, Brasília. Árvores, berinjela, vírus, resistência. I. Universidade de Brasília. PPG/FIT. II. Título. Ocorrência natural de espécies virais e avaliação de potenciais hospedeiras experimentais de begomovírus, potyvírus e tospovírus em plantas arbóreas e berinjela (Solanum melongena). Dedico a minha pequena linda filha Giovanna, ao meu pai Josvaldo, a minha mãe Nelma, ao meu namorado Airton, a minha irmã Josefa e minha sobrinha Júlia. Agradecimentos A Deus. Aos meus pais Josvaldo e Nelma. A minha filha Giovanna, minha irmã Josefa e minha sobrinha Júlia. A minha orientadora Professora Rita de Cássia. Aos colegas do mestrado e doutorado Rafaela, Marcella, Juliana, Ícaro, Cléia, Monica, Elenice, William, Rayane e Nédio. -

Identification of a Nanovirus-Alphasatellite Complex in Sophora Alopecuroides
Accepted Manuscript Title: Identification of a nanovirus-alphasatellite complex in Sophora alopecuroides Author: Jahangir Heydarnejad Mehdi Kamali Hossain Massumi Anders Kvarnheden Maketalena F. Male Simona Kraberger Daisy Stainton Darren P. Martin Arvind Varsani PII: S0168-1702(17)30009-6 DOI: http://dx.doi.org/doi:10.1016/j.virusres.2017.03.023 Reference: VIRUS 97113 To appear in: Virus Research Received date: 5-1-2017 Revised date: 15-3-2017 Accepted date: 18-3-2017 Please cite this article as: Heydarnejad, J., Kamali, M., Massumi, H., Kvarnheden, A., Male, M.F., Kraberger, S., Stainton, D., Martin, D.P., Varsani, A.,Identification of a nanovirus-alphasatellite complex in Sophora alopecuroides, Virus Research (2017), http://dx.doi.org/10.1016/j.virusres.2017.03.023 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. 1 Identification of a nanovirus-alphasatellite complex in Sophora alopecuroides 2 Jahangir Heydarnejad 1* , Mehdi Kamali 1, Hossain Massumi 1, Anders Kvarnheden 2, Maketalena 3 F. Male 3, Simona Kraberger 3,4 , Daisy Stainton 3,5 , Darren P. Martin 6 and Arvind Varsani 3,7,8* 4 5 1Department of Plant Protection, College