TRIM59 Interacts with ECSIT and Negatively Regulates NF-Κb and IRF-3/7-Mediated Signal Pathways
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Mechanism of ACAD9 in Mitochondrial Respiratory Complex 1 Assembly
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.07.425795; this version posted January 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Molecular mechanism of ACAD9 in mitochondrial respiratory complex 1 assembly Chuanwu Xia1, Baoying Lou1, Zhuji Fu1, Al-Walid Mohsen2, Jerry Vockley2, and Jung-Ja P. Kim1 1Department of Biochemistry, Medical College of Wisconsin, Milwaukee, Wisconsin, 53226, USA 2Department of Pediatrics, University of Pittsburgh School of Medicine, University of Pittsburgh, Children’s Hospital of Pittsburgh of UPMC, Pittsburgh, PA 15224, USA Abstract ACAD9 belongs to the acyl-CoA dehydrogenase family, which catalyzes the α-β dehydrogenation of fatty acyl-CoA thioesters. Thus, it is involved in fatty acid β-oxidation (FAO). However, it is now known that the primary function of ACAD9 is as an essential chaperone for mitochondrial respiratory complex 1 assembly. ACAD9 interacts with ECSIT and NDUFAF1, forming the mitochondrial complex 1 assembly (MCIA) complex. Although the role of MCIA in the complex 1 assembly pathway is well studied, little is known about the molecular mechanism of the interactions among these three assembly factors. Our current studies reveal that when ECSIT interacts with ACAD9, the flavoenzyme loses the FAD cofactor and consequently loses its FAO activity, demonstrating that the two roles of ACAD9 are not compatible. ACAD9 binds to the carboxy-terminal half (C-ECSIT), and NDUFAF1 binds to the amino-terminal half of ECSIT. Although the binary complex of ACAD9 with ECSIT or with C-ECSIT is unstable and aggregates easily, the ternary complex of ACAD9-ECSIT-NDUFAF1 (i.e., the MCIA complex) is soluble and extremely stable. -

TLR2 Signaling Pathway Combats Streptococcus Uberis Infection by Inducing Mitochondrial Reactive Oxygen Species Production
cells Article TLR2 Signaling Pathway Combats Streptococcus uberis Infection by Inducing Mitochondrial Reactive Oxygen Species Production 1, 1, 1 1,2 1 2 Bin Li y, Zhixin Wan y, Zhenglei Wang , Jiakun Zuo , Yuanyuan Xu , Xiangan Han , Vanhnaseng Phouthapane 3 and Jinfeng Miao 1,* 1 MOE Joint International Research Laboratory of Animal Health and Food Safty, Key Laboratory of Animal Physiology and Biochemistry, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing 210095, China; [email protected] (B.L.); [email protected] (Z.W.); [email protected] (Z.W.); [email protected] (J.Z.); [email protected] (Y.X.) 2 Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Shanghai 200241, China; [email protected] 3 Biotechnology and Ecology Institute, Ministry of Science and Technology (MOST), Vientiane 22797, Laos; [email protected] * Correspondence: [email protected]; Fax: +86-25-8439-8669 These authors contributed equally to this work. y Received: 6 January 2020; Accepted: 19 February 2020; Published: 21 February 2020 Abstract: Mastitis caused by Streptococcus uberis (S. uberis) is a common and difficult-to-cure clinical disease in dairy cows. In this study, the role of Toll-like receptors (TLRs) and TLR-mediated signaling pathways in mastitis caused by S. uberis was investigated using mouse models and mammary / epithelial cells (MECs). We used S. uberis to infect mammary glands of wild type, TLR2− − and / TLR4− − mice and quantified the adaptor molecules in TLR signaling pathways, proinflammatory cytokines, tissue damage, and bacterial count. When compared with TLR4 deficiency, TLR2 deficiency induced more severe pathological changes through myeloid differentiation primary response 88 (MyD88)-mediated signaling pathways during S. -

Ecsit Is Required for Bmp Signaling and Mesoderm Formation During Mouse Embryogenesis
Downloaded from genesdev.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Ecsit is required for Bmp signaling and mesoderm formation during mouse embryogenesis Changchun Xiao,1 Jae-hyuck Shim,1,6 Michael Klüppel,5,6 Samuel Shao-Min Zhang,3,6 Chen Dong,1,7 Richard A. Flavell,1 Xin-Yuan Fu,3 Jeffrey L. Wrana,5 Brigid L.M. Hogan,4 and Sankar Ghosh1,2,8 1Section of Immunobiology, 2Department of Molecular Biophysics and Biochemistry, Howard Hughes Medical Institute, and 3Department of Pathology, Yale University School of Medicine, New Haven, Connecticut 06520, USA; 4Department of Cell Biology, Duke University Medical Center, Durham, North Carolina 27710, USA; 5Programme in Molecular Biology and Cancer, Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, Ontario M5G 1X5, Canada Bone morphogenetic proteins (Bmps) are members of the transforming growth factor  (TGF) superfamily that play critical roles during mouse embryogenesis. Signaling by Bmp receptors is mediated mainly by Smad proteins. In this study, we show that a targeted null mutation of Ecsit, encoding a signaling intermediate of the Toll pathway, leads to reduced cell proliferation, altered epiblast patterning, impairment of mesoderm formation, and embryonic lethality at embryonic day 7.5 (E7.5), phenotypes that mimic the Bmp receptor type1a (Bmpr1a) null mutant. In addition, specific Bmp target gene expression is abolished in the absence of Ecsit. Biochemical analysis demonstrates that Ecsit associates constitutively with Smad4 and associates with Smad1 in a Bmp-inducible manner. Together with Smad1 and Smad4, Ecsit binds to the promoter of specific Bmp target genes. Finally, knock-down of Ecsit with Ecsit-specific short hairpin RNA inhibits both Bmp and Toll signaling. -

Mitochondrial Medicine in the Omics Era
Mitochondrial Medicine in the Omics Era Joyeeta Rahman1 and Shamima Rahman1,2* 1 Mitochondrial Research Group, UCL Great Ormond Street Institute of Child Health and 2 Metabolic Unit, Great Ormond Street Hospital NHS Foundation Trust, London, UK *Correspondence to: Professor Shamima Rahman Mitochondrial Research Group Genetics and Genomic Medicine UCL Great Ormond Street Institute of Child Health London WC1N 1EH, UK. Telephone: +44 (0)2079052608 [email protected] Keywords: Mitochondrial disease, OXPHOS, signalling, omics, genomics, transcriptomics, proteomics, metabolomics, mitochondrial stress response, treatment 1 Abstract Mitochondria are dynamic bioenergetic organelles whose maintenance requires ~1500 proteins from two genomes. Mutations in either the mitochondrial or nuclear genome can disrupt a plethora of cellular metabolic and homeostatic functions. Mitochondrial diseases represent one the most common and severe groups of inherited genetic disorders, characterised by clinical, biochemical, and genetic heterogeneity, diagnostic odysseys, and lack of curative therapies. This review aims to discuss recent advances in mitochondrial biology and medicine arising from widespread utilisation of high-throughput omics technologies, and also includes a broad discussion of emerging therapies for mitochondrial disease. New insights into both bioenergetic and biosynthetic mitochondrial functionalities have expedited the genetic diagnosis of primary mitochondrial disorders, and identified novel mitochondrial pathomechanisms and new targets -

6 Signaling and BMP Antagonist Noggin in Prostate Cancer
[CANCER RESEARCH 64, 8276–8284, November 15, 2004] Bone Morphogenetic Protein (BMP)-6 Signaling and BMP Antagonist Noggin in Prostate Cancer Dominik R. Haudenschild, Sabrina M. Palmer, Timothy A. Moseley, Zongbing You, and A. Hari Reddi Center for Tissue Regeneration and Repair, Department of Orthopedic Surgery, School of Medicine, University of California, Davis, Sacramento, California ABSTRACT antagonists has recently been discovered. These are secreted proteins that bind to BMPs and reduce their bioavailability for interactions It has been proposed that the osteoblastic nature of prostate cancer with the BMP receptors. Extracellular BMP antagonists include nog- skeletal metastases is due in part to elevated activity of bone morphoge- gin, follistatin, sclerostatin, chordin, DCR, BMPMER, cerberus, netic proteins (BMPs). BMPs are osteoinductive morphogens, and ele- vated expression of BMP-6 correlates with skeletal metastases of prostate gremlin, DAN, and others (refs. 11–16; reviewed in ref. 17). There are cancer. In this study, we investigated the expression levels of BMPs and several type I and type II receptors that bind to BMPs with different their modulators in prostate, using microarray analysis of cell cultures affinities. BMP activity is also regulated at the cell membrane level by and gene expression. Addition of exogenous BMP-6 to DU-145 prostate receptor antagonists such as BAMBI (18), which acts as a kinase- cancer cell cultures inhibited their growth by up-regulation of several deficient receptor. Intracellularly, the regulation of BMP activity at cyclin-dependent kinase inhibitors such as p21/CIP, p18, and p19. Expres- the signal transduction level is even more complex. There are inhib- sion of noggin, a BMP antagonist, was significantly up-regulated by itory Smads (Smad-6 and Smad-7), as well as inhibitors of inhibitory BMP-6 by microarray analysis and was confirmed by quantitative reverse Smads (AMSH and Arkadia). -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

Instability in NAD Metabolism Leads to Impaired Cardiac Mitochondrial
RESEARCH ARTICLE Instability in NAD+ metabolism leads to impaired cardiac mitochondrial function and communication Knut H Lauritzen1*, Maria Belland Olsen1, Mohammed Shakil Ahmed2, Kuan Yang1, Johanne Egge Rinholm3, Linda H Bergersen4,5, Qin Ying Esbensen6, Lars Jansen Sverkeli7, Mathias Ziegler7, Ha˚ vard Attramadal2, Bente Halvorsen1,8, Pa˚ l Aukrust1,8,9, Arne Yndestad1,8 1Research Institute of Internal Medicine, Oslo University Hospital, Rikshospitalet and University of Oslo, Oslo, Norway; 2Institute for Surgical Research, Oslo University Hospital and University of Oslo, Oslo, Norway; 3Department of Microbiology, Oslo University Hospital, Oslo, Norway; 4Department of Oral Biology, University of Oslo, Oslo, Norway; 5Department of Neuroscience and Pharmacology, Center for Healthy Aging, University of Copenhagen, Copenhagen, Denmark; 6Department of Clinical Molecular Biology, University of Oslo and Akershus University Hospital, Nordbyhagen, Norway; 7Department of Biomedicine, University of Bergen, Bergen, Norway; 8Institute of Clinical Medicine, University of Oslo, Faculty of Medicine, Oslo, Norway; 9Section of Clinical Immunology and Infectious Diseases, Oslo University Hospital Rikshospitalet, Oslo, Norway Abstract Poly(ADP-ribose) polymerase (PARP) enzymes initiate (mt)DNA repair mechanisms and use nicotinamide adenine dinucleotide (NAD+) as energy source. Prolonged PARP activity can drain cellular NAD+ reserves, leading to de-regulation of important molecular processes. Here, we provide evidence of a pathophysiological mechanism that connects mtDNA damage to cardiac *For correspondence: dysfunction via reduced NAD+ levels and loss of mitochondrial function and communication. Using Knut.Huso.Lauritzen@rr-research. a transgenic model, we demonstrate that high levels of mice cardiomyocyte mtDNA damage cause no a reduction in NAD+ levels due to extreme DNA repair activity, causing impaired activation of Competing interests: The NAD+-dependent SIRT3. -

Supporting Information
Supporting Information Noyes et al. 10.1073/pnas.1013486108 Table S1. Abbreviations Gene name Description ADRB3 Adrenergic, β-3-, receptor AGPAT6 1-acylglycerol-3-phosphate O-acyltransferase 6 (lysophosphatidic acid acyltransferase, zeta) ARHGAP15 Rho GTPase activating protein 15 BRAF v-raf murine sarcoma viral oncogene homolog B1 CAPN2 Calpain 2, (m/II) large subunit CASP2 Caspase 2, apoptosis-related cysteine peptidase CASP8 Caspase 8, apoptosis-related cysteine peptidase CD14 CD14 molecule CD28 CD28 molecule CDKN2D Cyclin-dependent kinase inhibitor 2D (p19, inhibits CDK4) CHRM2 Cholinergic receptor, muscarinic 2 CHRM2 Cholinergic receptor, muscarinic 2 CLDN23 Claudin 23 CTLA4 Cytotoxic t lymphocyte-associated protein 4 DGKI Diacylglycerol kinase, iota DKK4 Dickkopf homolog 4 (xenopus laevis) DUSP10 Dual-specificity phosphatase 10 DUSP4 Dual-specificity phosphatase 4 ECSIT ECSIT homolog (Drosophila) ECSIT ECSIT homolog (Drosophila) EPHA1 EPH receptor A1 EPHB6 EPH receptor B6 EPOR Erythropoietin receptor FCER2 Fc fragment of IgE, low affinity II, receptor for (CD23) FGF20 Fibroblast growth factor 20 FGFR1 Fibroblast growth factor receptor 1 GNA14 Guanine nucleotide binding protein (G protein), alpha 14 GNAQ Guanine nucleotide binding protein (G protein), q polypeptide GSTK1 GST kappa 1 ICAM1 Intercellular adhesion molecule 1 ICAM3 Intercellular adhesion molecule 3 ICOS Inducible T-cell costimulator IDH1 Isocitrate dehydrogenase 1 (nadp+), soluble IKBKB Inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase beta INSR Insulin -

Anti-ECSIT Monoclonal Antibody (DCABH-15245) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-ECSIT monoclonal antibody (DCABH-15245) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Antigen Description Adapter protein of the Toll-like and IL-1 receptor signaling pathway that is involved in the activation of NF-kappa-B via MAP3K1. Promotes proteolytic activation of MAP3K1. Involved in the BMP signaling pathway. Required for normal embryonic development (By similarity). Required for efficient assembly of mitochondrial NADH:ubiquinone oxidoreductase. Immunogen A synthetic peptide of human ECSIT is used for rabbit immunization. Isotype IgG Source/Host Rabbit Species Reactivity Human Purification Protein A Conjugate Unconjugated Applications Western Blot (Transfected lysate); ELISA Size 1 ea Buffer In 1x PBS, pH 7.4 Preservative None Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. GENE INFORMATION Gene Name ECSIT ECSIT homolog (Drosophila) [ Homo sapiens ] Official Symbol ECSIT Synonyms ECSIT; ECSIT homolog (Drosophila); evolutionarily conserved signaling intermediate in Toll pathway, mitochondrial; signaling intermediate in Toll pathway evolutionarily conserved ortholog (mouse); SITPEC; likely ortholog of mouse signaling intermediate in Toll pathway evolutionarily conserved; Entrez Gene ID 51295 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Protein Refseq NP_001135936 UniProt ID Q9BQ95 Chromosome Location 19p13.2 Pathway Activated TLR4 signalling, organism-specific biosystem; Immune System, organism-specific biosystem; Innate Immune System, organism-specific biosystem; MAPK signaling pathway, organism-specific biosystem; MAPK signaling pathway, organism-specific biosystem Function oxidoreductase activity, acting on NADH or NADPH; protein binding; 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 2 © Creative Diagnostics All Rights Reserved. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Conceptual Evolution of Cell Signaling
International Journal of Molecular Sciences Review Conceptual Evolution of Cell Signaling 1, 1, 1, 2, Arathi Nair y , Prashant Chauhan y , Bhaskar Saha * and Katharina F. Kubatzky * 1 National Center for Cell Science (NCCS), Ganeshkhind, Pune 411007, India 2 Zentrum für Infektiologie, Medizinische Mikrobiologie und Hygiene, Universitätsklinikum Heidelberg, Im Neuenheimer Feld 324, 69120 Heidelberg, Germany * Correspondence: [email protected] (B.S.); [email protected] (K.F.K.) Indicates Equal Contribution. y Received: 11 April 2019; Accepted: 28 June 2019; Published: 4 July 2019 Abstract: During the last 100 years, cell signaling has evolved into a common mechanism for most physiological processes across systems. Although the majority of cell signaling principles were initially derived from hormonal studies, its exponential growth has been supported by interdisciplinary inputs, e.g., from physics, chemistry, mathematics, statistics, and computational fields. As a result, cell signaling has grown out of scope for any general review. Here, we review how the messages are transferred from the first messenger (the ligand) to the receptor, and then decoded with the help of cascades of second messengers (kinases, phosphatases, GTPases, ions, and small molecules such as cAMP, cGMP, diacylglycerol, etc.). The message is thus relayed from the membrane to the nucleus where gene expression ns, subsequent translations, and protein targeting to the cell membrane and other organelles are triggered. Although there are limited numbers of intracellular messengers, the specificity of the response profiles to the ligands is generated by the involvement of a combination of selected intracellular signaling intermediates. Other crucial parameters in cell signaling are its directionality and distribution of signaling strengths in different pathways that may crosstalk to adjust the amplitude and quality of the final effector output. -
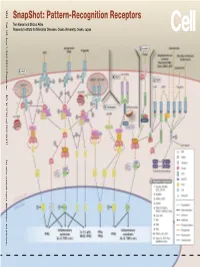
Snapshot: Pattern-Recognition Receptors
SnapShot: Pattern-Recognition Receptors SnapShot:Pattern-Recognition Kawai and Shizuo Akira Taro Osaka, Japan Diseases, Osaka University, Institute for Microbial Research 1024 Cell 129, June 1, 2007 ©2007 Elsevier Inc. DOI 10.1016/j.cell.2007.05.017 See online version for legend, abbreviations, and references. SnapShot: Pattern-Recognition Receptors Taro Kawai and Shizuo Akira Research Institute for Microbial Diseases, Osaka University, Osaka, Japan (A) Toll-like receptor signaling. Toll-like receptor (TLR) 3 recognizes polyinosinic-polycytidylic acid (poly IC), whereas TLR4 recognizes lipopolysaccharide (LPS). TLR2 recog- nizes various components such as lipoprotein and peptidoglycan (PGN). TLR5 detects flagellin. TLR7 and TLR9 detect single-stranded (ss)RNA and CpG DNA, respectively. Each TLR recruits a distinct set of Toll/interleukin-1 receptor (TIR) domain-containing adaptor molecules such as myeloid differentiation primary response gene 88 (MyD88), TIR-containing adaptor protein (TIRAP, also known as MAL), TIR-containing adaptor-inducing IFNβ (TRIF, also known as TICAM1) and TRIF-related adaptor molecule (TRAM, also known as TICAM2). TLR3 uses TRIF, and TLR5, 7, and 9 use MyD88. TLR2 uses MyD88 and TIRAP, and TLR4 uses MyD88, TIRAP, TRIF, and TRAM. MyD88 binds to inter- leukin-1 receptor-associated kinase 4 (IRAK4) and TRAF6. TRIF binds receptor-interacting protein 1 (RIP1) and TRAF6. TRAF6 forms a complex with Ubc13, Uev1A, and ECSIT (evolutionarily conserved signaling intermediate in Toll/IL-1R pathways) to activate a complex containing transforming growth factor-β-activated kinase 1 (TAK1), TAK1-binding protein 1 (TAB1), TAB2, and TAB3. TAK1 activates IκB kinase (IKK) complex consisting of IKKα, IKKβ, and Nemo (also known as IKKγ), which results in the phosphorylation and proteasomal degradation of IκB proteins and the release of a transcription factor NFκB to the nucleus to regulate expression of inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor α (TNFα).