Total Synthesis of Verruculogen
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Professor of Chemistry
Bruce C. Gibb FRSC Professor of Chemistry Department of Chemistry Tulane University New Orleans, LA 70118, USA Tel: (504) 862 8136 E-mail: [email protected] Website: http://www.gibbgroup.org Twitter: @brucecgibb Research Interests: Aqueous supramolecular chemistry: understanding how molecules interact in water: from specific ion- pairing and the hydrophobic effect, to protein aggregation pertinent to neurodegenerative disorders. Our research has primarily focused on: 1) novel hosts designed to probe the hydrophobic, Hofmeister, and Reverse Hofmeister effects, and; 2) designing supramolecular capsules as yocto-liter reaction vessels and separators. Current efforts to probe the hydrophobic and Hofmeister effects include studies of the supramolecular properties of proteins. Professional Positions: Visiting Professor, Wuhan University of Science and Technology as a Chair Professor of Chutian Scholars Program (2015-2018) Professor of Chemistry, Tulane University, New Orleans, USA (2012-present). University Research Professor, University of New Orleans, USA (2007-2011). Professor of Chemistry, University of New Orleans, USA (2005-2007). Associate Professor of Chemistry, University of New Orleans, USA (2002-2005). Assistant Professor of Chemistry, University of New Orleans, USA, (1996-2002). Education: Postdoctoral Work Department of Chemistry, New York University. Synthesis of Carbonic Anhydrase (CA) mimics with Advisor: Prof. J. W. Canary, (1994-1996). Department of Chemistry, University of British Columbia, Canada De Novo Protein development. Advisor: Prof. J. C. Sherman (1993-1994). Ph.D. Robert Gordon’s University, Aberdeen, UK. Synthesis and Structural Examination of 3a,5-cyclo-5a- Androstane Steroids. Advisors: Dr. Philip J. Cox and Dr. Steven MacManus (1987-92) . B.Sc. with Honors in Physical Sciences Robert Gordon’s University, Aberdeen, UK. -

Molecular Electronic Devices Based on Single-Walled Carbon Nanotube Electrodes Alina K
Subscriber access provided by Columbia Univ Libraries Article Molecular Electronic Devices Based on Single-Walled Carbon Nanotube Electrodes Alina K. Feldman, Michael L. Steigerwald, Xuefeng Guo, and Colin Nuckolls Acc. Chem. Res., 2008, 41 (12), 1731-1741• DOI: 10.1021/ar8000266 • Publication Date (Web): 18 September 2008 Downloaded from http://pubs.acs.org on May 14, 2009 More About This Article Additional resources and features associated with this article are available within the HTML version: • Supporting Information • Access to high resolution figures • Links to articles and content related to this article • Copyright permission to reproduce figures and/or text from this article Accounts of Chemical Research is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036 Molecular Electronic Devices Based on Single-Walled Carbon Nanotube Electrodes ALINA K. FELDMAN,† MICHAEL L. STEIGERWALD,† XUEFENG GUO,*,‡ AND COLIN NUCKOLLS*,† †Department of Chemistry and the Columbia University Center for Electronics of Molecular Nanostructures, Columbia University, New York, New York 10027, ‡Centre for Nanochemistry (CNC), Beijing National Laboratory for Molecular Sciences (BNLMS), State Key Laboratory for Structural Chemistry of Unstable and Stable Species, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China RECEIVED ON JANUARY 28, 2008 CON SPECTUS s the top-down fabrication techniques for silicon-based Aelectronic materials have reached the scale of molecu- lar lengths, researchers have been investigating nanostruc- tured materials to build electronics from individual molecules. Researchers have directed extensive experimental and the- oretical efforts toward building functional optoelectronic devices using individual organic molecules and fabricating metal-molecule junctions. Although this method has many advantages, its limitations lead to large disagreement between experimental and theoretical results. -

Jacques Lux, Phd
CURRICULUM VITAE Jacques Lux, PhD PERSONAL INFORMATION Name: Jacques Lux, PhD Place of Birth: Le Mans, France Year Of Birth: 1982 Home Address: 3930 McKinney Avenue Apt. 329 Dallas, TX 75204 Home Phone: Office Address: 5323 Harry Hines Blvd. Dallas, TX 75390-8514 Office Phone: 214-648-5091 Fax: 214-648-5097 Office Email: [email protected] EDUCATION Year Degree Field of Study Institution 2004 BSc Chemistry and Physics University of Upper Alsace (France) 2006 Engineer Organic Chemistry Mulhouse National School of Chemistry (France) 2006 MSc Chemistry University of Upper Alsace (France) 2009 PhD Chemistry University of Strasbourg, Institute of Chemistry (France) POSTDOCTORAL TRAINING Year(s) Training Specialty/Discipline Institution 2009-2010 Fellowship Fluorescent Probes and University of Strasbourg, Faculty of Pharmacy (France) Two-photon Microscopy 2010-2012 Fellowship Supramolecular Chemistry, The Scripps Research Institute Host-guest Chemistry 2012-2014 Fellowship Bioresponsive Materials, University of California San Diego, San Diego, CA MRI/PET Contrast Agents FACULTY ACADEMIC APPOINTMENT Year(s) Academic Title Academic Department Academic Institution 2015-now Assistant Professor Radiology University of Texas Southwestern Medical School, Dallas, TX 2016-2017 Assistant Professor Biological Chemistry University of Texas Southwestern Medical School, Dallas, TX Graduate Program 2016-now Assistant Professor Organic Chemistry Graduate University of Texas Southwestern Medical School, Dallas, TX Program 2017-now Assistant Professor -

Frontiers in Chemistry Lecture Series Sponsored by Frontiers in Chemistry Endowment
Frontiers in Chemistry Lecture Series Sponsored by Frontiers in Chemistry Endowment Development of Proof-of-Concept Chemical Probes Targeting Novel Biological Targets Professor William R. Roush Scripps Research Institute, Florida 4:00 p.m. Friday, October 4th, 2013 WO 1201 William R. Roush Professor and Executive Director of Medicinal Chemistry and Associate Dean of Scripps' Kellogg Graduate School in Jupiter. Florida Dr. William R. Roush, a native of Chula Vista, CA, received the Bachelors Degree in Chemistry, Summa Cum Laude, from the University of California at Los Angeles in 1974, where he performed undergraduate research with Professor Julius Rebek, and the Ph. D. Degree in Chemistry from Harvard University in 1977 under the direction of Professor R. B. Woodward. After an additional year as a postdoctoral associate in Woodward's laboratory, he joined the Massachusetts Institute of Technology as Assistant Professor. He moved to Indiana University in 1987, and was promoted to the rank of Professor in 1989 and Distinguished Professor in 1995. In 1997 he moved to the University of Michigan as the Warner Lambert/Parke Davis Professor of Chemistry. He served as Chair of the Department of Chemistry, University of Michigan, from 2002-2004. He moved to the new Scripps Research Institute in Jupiter, Florida, as Professor of Chemistry, Executive Director of Medicinal Chemistry, and Associate Dean of Scripps’ Kellogg Graduate School in 2005. Dr. Roush has been a Fellow of the Alfred P. Sloan Foundation, an Eli Lilly Grantee, and the holder of the Roger and Georges Firmenich Career Development Chair in Natural Products Chemistry at MIT. -
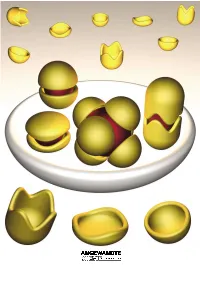
Molecular Encapsulation
REVIEWS Molecular Encapsulation Fraser Hof, Stephen L. Craig, Colin Nuckolls, and Julius Rebek, Jr.* Dedicated to Ivar Ugi Louis Kahn, architect of the Salk nents. This review is about molecular ble–an expression of the molecule×s Institute in La Jolla, said[1] ™even a aggregates of a certain sort, namely, desire to be something more than it common, ordinary brick wants to be those that assemble and more or less is–results from instructions engi- something more than it is.∫ Suppose completely surround other molecules. neered into the molecules during their that were also true of molecules. We Taking part in this intimacy imparts creation. know that they can and do aggregate; unique properties to the participants, they give complex structures, and by and new functions emerge from the Keywords: host ± guest systems ¥ mo- doing so they acquire new properties– aggregate as a whole. For the most lecular capsules ¥ molecular recogni- functions that may not be apparent part, we emphasize self-complementa- tion ¥ self-assembly from a study of the individual compo- ry structures. Their ability to assem- 1. Introduction reaction templates were devised[17] and more complex systems were contrived. This is engineering (or is it art?)[18] at the In the 1980s, most of the publications on molecular molecular level.[19] It did not matter that these systems weren×t recognition dealt with the selectivity of synthetic receptors particularly efficient, what mattered was that they improved and the energetics of intermolecular forces, and were confined the understanding of three-component systems. for the most part to bimolecular systems. -

Slayton A. Evans, Jr
The Slayton A. Evans, Jr. Lecture in Chemistry Slayton A. Evans, Jr. Slayton Evans was the kind of person professors ought to be; a genial mentor and advisor to his students, a first-rate researcher, and a special friend to his colleagues. Born in Mississippi in 1943, Evans’ studies started at Tougaloo College and included graduate work at Case Western Reserve, and postdoctoral work at University of Texas at Arlington and Notre Dame, before he joined the faculty at Carolina in 1974. He became the chemistry department’s first African-American professor. Evans’ commitment to improve science education for future generations was as significant as his being a successful academic scientist in one of the nation’s top universities. Locally, as a member of the Pogue Undergradu- ate Scholarship Committee, he persuaded the chancellor to augment both the number and value of scholarships substantially and to direct them to the promotion of diversity in the undergraduate student body. Nationally, he was a valued policy advisor to both the National Science Foundation and the National Institutes of Health. In addition to his contributions within his own specialty of organophosphorus chemistry, Evans used his intellect and integrity to make sure other minorities also found their place in Carolina’s chemistry program. His colleagues and students repeatedly praised Evans for having as much insight about how to treat people as he did about conducting his science. This ability to teach his students about science as well as how to excel professionally gar- nered Evans the respect of many who found his pedagogical approach inspiring and worthy of numerous awards and accolades. -

Julius Rebek, Jr
JULIUS REBEK, JR. Director, The Skaggs Institute for Chemical Biology and Professor of Chemistry, Department of Chemistry The Scripps Research Institute Biographical Sketch Julius Rebek, Jr. was born in Hungary in 1944 and lived in Austria from 1945-49. He and his family then settled in the U.S.A. in Kansas. He received his undergraduate education at the University of Kansas in 1966, and obtained the Ph.D. degree from the Massachusetts Institute of Technology (1970) for studies in peptide chemistry with Professor D.S. Kemp. As an Assistant Professor at the University of California at Los Angeles (1970-1976) he developed the three- phase test for reactive intermediates. In 1976 he moved to the University of Pittsburgh where he rose to the rank of Professor of Chemistry and developed cleft-like structures for studies in molecular recognition. In 1989 he returned to the Massachusetts Institute of Technology, where he was the Camille Dreyfus Professor of Chemistry and devised synthetic, self-replicating molecules. In July of 1996, he moved his research group to The Scripps Research Institute to become the Director of The Skaggs Institute for Chemical Biology, where he continues to work in molecular recognition and self-assembling systems. Biographical Data Birth date: April 11, 1944; Beregszasz, Hungary Education: B.A. University of Kansas, 1966 Ph.D. Massachusetts Institute of Technology, 1970 Positions: University of California, Los Angeles Assistant Professor, 1970-1976 University of Pittsburgh Associate Professor, 1976-1979 Professor, 1980-1989 Massachusetts Institute of Technology, Cambridge, MA Professor, 1989-1991 Camille Dreyfus Professor of Chemistry, 1991-1996 The Scripps Research Institute, La Jolla, CA Director, The Skaggs Institute for Chemical Biology and Professor of Chemistry, 1996 - 1 - Honors and Awards NSF Predoctoral Fellow, 1967-1970 Eli Lilly Award, 1972-1974 A. -

American Chemical Society Division of Organic Chemistry 243Rd ACS National Meeting, San Diego, CA, March 25-29, 2012
American Chemical Society Division of Organic Chemistry 243rd ACS National Meeting, San Diego, CA, March 25-29, 2012 A. Abdel-Magid, Program Chair; R. Gawley, Program Chair SUNDAY MORNING Ralph F. Hirschmann Award in Peptide Chemistry: Symposium in Honor of Jeffery W. Kelly D. Huryn, Organizer; D. Huryn, Presiding Papers 1-4 Biologically-Related Molecules and Processes A. Abdel-Magid, Organizer; T. Altel, Presiding Papers 5-16 New Reactions and Methodology A. Abdel-Magid, Organizer; N. Bhat, Presiding Papers 17-28 Asymmetric Reactions and Syntheses A. Abdel-Magid, Organizer; D. Leahy, Presiding Papers 29-39 Material, Devices, and Switches A. Abdel-Magid, Organizer; S. Thomas, Presiding Papers 40-49 SUNDAY AFTERNOON James Flack Norris Award in Physical Organic Chemistry: Symposium to Honor Hans J. Reich G. Weisman, Organizer; G. Weisman, Presiding Papers 50-53 Understanding Additions to Alkenes D. Nelson, Organizer; D. Nelson, Presiding Papers 54-61 Biologically-Related Molecules and Processes A. Abdel-Magid, Organizer; B. C. Das, Presiding Papers 62-73 New Reactions and Methodology A. Abdel-Magid, Organizer; T. Minehan, Presiding Papers 74-85 Asymmetric Reactions and Syntheses A. Abdel-Magid, Organizer; A. Mattson, Presiding Papers 86-97 Material, Devices, and Switches A. Abdel-Magid, Organizer; Y. Cui, Presiding Papers 98-107 SUNDAY EVENING Material, Devices, and Switches, Molecular Recognition, Self-Assembly, Peptides, Proteins, Amino Acids, Physical Organic Chemistry, Total Synthesis of Complex Molecules R. Gawley, Organizer Papers 108-257 MONDAY MORNING Herbert C. Brown Award for Creative Research in Synthetic Organic Chemistry: Symposium in Honor of Jonathan A. Ellman S. Sieburth, Organizer; S. Sieburth, Presiding Papers 258-261 Playing Ball: Molecular Recognition and Modern Physical Organic Chemistry C. -

Salk Bulletin Extra
Bulletin From: Bulletin <[email protected]> Sent: Thursday, September 28, 2017 12:15 PM To: '[email protected]' Subject: Salk Bulletin, October 2, 2017 - October 9, 2017 Salk Bulletin Monday, October 2, 2017 Monday, October 2, 2017 9:30 am “T Memory Stem Cells in Health and Disease: New Insights and Therapeutic Opportunities” Luca Gattinoni, M.D., Stadtman Investigator, National Cancer Institute , National Institutes of Health Immunobiology Seminar Series UNIVERSITY OF CALIFORNIA SAN DIEGO 1205 Natural Sciences Building Monday, October 2, 2017 12:00 pm “Autophagy, Cell Death and Cancer Therapy” Dr. Andrew Thorburn, University of Colorado Cancer Center Cancer Center Seminar Series SANFORD BURNHAM PREBYS MEDICAL DISCOVERY INSTITUTE Fishman Auditorium Host: Malene Hansen Contact: Sarah Forte ext. 5023 1 Monday, October 2, 2017 5:00 - 6:00 pm “Single-Cell Methylome Sequencing of Identified Cortical Projection Cell Types” Dr. Zhuzhu Zhang, Joseph Ecker Lab, Salk Institute for Biological Studies “Characterization of VEGF-Independent Angiogenesis in a HIF1A- Knockdown Model” Marco Maruggi, M. Eng., Ph.D. Candidate; NIH NRSA Predoctoral Fellow, Dr. Garth Powis Lab, Sanford Burnham Prebys Medical Discovery Institute C3-SCS (Single Cell Sequencing) Workshop Group/Social SANFORD BURNHAM PREBYS MEDICAL DISCOVERY INSTITUTE Fishman Auditorium Hosts: Drs. Peter Adams, Brian James and Geoffrey Wahl No Seminar Listings At This Time Tuesday, October 3, 2017 Tuesday, October 3, 2017 9:00 - 10:00 am Clinicopathologic Comference (CPC) Cases will be presented -

ABCD Just Released New Books February 2012
ABCD springer.com Just Released New Books February 2012 English Titles Sorted by author and title within the main subject springer.com Architecture & Design 2 Architecture & Design Arts Biomedicine J. Portugali, Tel Aviv University,Israel; H. Meyer, TU Delft, H. Selin, Hampshire College, 893 West St, Amherts, MA 01002, R. Scatena, Catholic University, Rome, Italy; P. Bottoni, Catholic Netherlands; E. Stolk, TU Delft, Netherlands; E. Tan, TU Delft, USA; G. Davey, Hong Kong Shue Yan University, China (Eds.) University, Rome, Italy; B. Giardina, Catholic University, Rome, Netherlands (Eds.) Italy (Eds.) Happiness Across Cultures Complexity Theories of Cities Have Advances in Mitochondrial Views of Happiness and Quality of Life in Non-Western Come of Age Cultures Medicine An Overview with Implications to Urban Planning and Design Different cultures experience happiness differently. Mitochondria are far more than the “powerhouse” Traditionally, the West is considered materialistic, Today, our cities are an embodiment of the complex, of the cell as they have classically been described. and happiness is said to come from achievement historical evolution of knowledge, desires and In fact, mitochondria biological activities have and acquisition. The East is said to be more people- technology. Our planned and designed activities progressively expanded to include not only oriented, where happiness is a result of deep personal co-evolve with our aspirations, mediated by the various bioenergetic processes but also important interactions. Thus, poor people can be happier in existing technologies and social structures. The biosynthetic pathways, calcium homeostasis and the East than the West, because they are not so city represents the accretion and accumulation of thermogenesis, cell death by apoptosis, several concerned with possession and more with society. -

Program for the 36Th
· University of Wisconsin, Madison, Wisconr~ d~ 36th National Organic Chemistry Symposium June 13 -17,1999 University of Wisconsin Madison, Wisconsin Table of Contents Program ........................................................................................................ ii Symposium Organizers and Divisional Officers ......................................... vi ACS Organic Division Graduate Fellows and Sponsors ............................ vii Roger Adams Award ............. __ ...................................................................... x Campus Map ................................................................................................ xi Memorial Union Floor Layout ................................................................... xii Lecture Abstracts Elias J. Corey .................................................................................... 1. Sheila De "Witt ..................................................................................... 7 Gregory Fu ...................................................................................... 15 Samuel Gellman ............................................................................... 20 Robert Grubbs ................................................................................. 25 Chaitan Kholsa ................................................................................ 32 Dieter Seebach ................................................................................. 36 Jeffrey Moore .................................................................................. -

Seminars 2005-06
CALIFORNIA INSTITUTE OF TECHNOLOGY SEMINARS IN CHEMISTRY AND CHEMICAL ENGINEERING October 6, 2006 BIS Beckman Institute Seminar IES Inorganic-Electrochemistry Seminar BL Beckman Lecture IOS Inorganic-Organometallics Seminar BLS Biophysics Lecture Series JDRL John D. Roberts Lecture BMSLOS Bristol-Myers Squibb Lectures in Organic Synthesis LMS Laboratory for Molecular Sciences Seminar BS Biochemistry Seminar LPL Linus Pauling Lectureship CBC Caltech Biotech Club MAS McCoy Award Seminar CCS Chemistry Club Seminar MRL Materials Research Lecture (CCE co-sponsor) CES Chemical Engineering Seminar NDL Norman Davidson Lecture CGEL Constantin G. Economou Memorial Lecture OCS Organic Chemistry Seminar CPS Chemical Physics Seminar RWVL Robert W. Vaughan Lectureship in Chem. Eng. CRC Chemical Research Conference SL Swift Lecture DDS Doctoral Dissertation Seminar SS Special Seminar DLOMC Dow Lecture in Organometallic Chemistry WNLL W.N. Lacey Lectureship in Chem. Eng. DLOC Dow Lecture in Organic Chemistry Type Date Time Location Speaker Title Ext. IOS 10/06 4:00 PM 151 Crellin Dr. Patricio Romero “14-Electron Ruthenium 6022 Caltech Postdoctoral Scholar Phosphonium Alkylidenes in (Grubbs Research Group) Olefin Metathesis” SS 10/11 4:00 PM 153 Noyes Prof. Donna G. Blackmond “Exploring the Origin of 6151 Imperial College Biological Homochirality: How Catalysis May Have Played a Role” IOS 10/13 4:00 PM 151 Crellin Dr. Michael Malarek “Novel π-Acid-Base Chemistry 6022 Caltech Postdoctoral Scholar of Molybdenum” (Grubbs Research Group) OCS 10/18 4:00 PM 153 Noyes Prof. Matthew B. Francis “New Chemical Tools for 6151 University of California, at Site-Selective Protein Berkeley Modification” BS 10/19 4:00 PM 147 Noyes Prof.