123.312 Advanced Organic Chemistry: Retrosynthesis Tutorial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Philosophy of Science and Philosophy of Chemistry
Philosophy of Science and Philosophy of Chemistry Jaap van Brakel Abstract: In this paper I assess the relation between philosophy of chemistry and (general) philosophy of science, focusing on those themes in the philoso- phy of chemistry that may bring about major revisions or extensions of cur- rent philosophy of science. Three themes can claim to make a unique contri- bution to philosophy of science: first, the variety of materials in the (natural and artificial) world; second, extending the world by making new stuff; and, third, specific features of the relations between chemistry and physics. Keywords : philosophy of science, philosophy of chemistry, interdiscourse relations, making stuff, variety of substances . 1. Introduction Chemistry is unique and distinguishes itself from all other sciences, with respect to three broad issues: • A (variety of) stuff perspective, requiring conceptual analysis of the notion of stuff or material (Sections 4 and 5). • A making stuff perspective: the transformation of stuff by chemical reaction or phase transition (Section 6). • The pivotal role of the relations between chemistry and physics in connection with the question how everything fits together (Section 7). All themes in the philosophy of chemistry can be classified in one of these three clusters or make contributions to general philosophy of science that, as yet , are not particularly different from similar contributions from other sci- ences (Section 3). I do not exclude the possibility of there being more than three clusters of philosophical issues unique to philosophy of chemistry, but I am not aware of any as yet. Moreover, highlighting the issues discussed in Sections 5-7 does not mean that issues reviewed in Section 3 are less im- portant in revising the philosophy of science. -

3-Monochloropropane-1,2-Diol Esters and Glycidyl Esters
www.nature.com/scientificreports OPEN Monitoring of heat‑induced carcinogenic compounds (3‑monochloropropane‑1,2‑diol esters and glycidyl esters) in fries Yu Hua Wong1, Kok Ming Goh1,2, Kar Lin Nyam2, Ling Zhi Cheong3, Yong Wang4, Imededdine Arbi Nehdi5,6, Lamjed Mansour7 & Chin Ping Tan1* 3‑Monochloropropane‑1,2‑diol (3‑MCPD) esters and glycidyl esters (GE) are heat‑induced contaminants which form during oil refning process, particularly at the high temperature deodorization stage. It is worth to investigate the content of 3‑MCPD and GE in fries which also involved high temperature. The content of 3‑MCPD esters and GE were monitored in fries. The factors that been chosen were temperature and duration of frying, and diferent concentration of salt (NaCl). The results in our study showed that the efect was in the order of concentration of sodium chloride < frying duration < frying temperature. The content of 3‑MCPD esters was signifcantly increased whereas GE was signifcantly decreased, when prolong the frying duration. A high temperature results in a high 3‑MCPD ester level but a low GE level in fries. The present of salt had contributed signifcant infuence to the generation of 3‑MCPD. The soaking of potato chips in salt showed no signifcant efect on the level of GE during the frying. The oil oxidation tests showed that all the fries were below the safety limit. Hence, the frying cycle, temperature and the added salt to carbohydrate‑based food during frying should be monitored. Deep-fat frying is commonly being used to process food. During the process, heat transfer between the fried food and oil is occurs. -

Peptide Chemistry up to Its Present State
Appendix In this Appendix biographical sketches are compiled of many scientists who have made notable contributions to the development of peptide chemistry up to its present state. We have tried to consider names mainly connected with important events during the earlier periods of peptide history, but could not include all authors mentioned in the text of this book. This is particularly true for the more recent decades when the number of peptide chemists and biologists increased to such an extent that their enumeration would have gone beyond the scope of this Appendix. 250 Appendix Plate 8. Emil Abderhalden (1877-1950), Photo Plate 9. S. Akabori Leopoldina, Halle J Plate 10. Ernst Bayer Plate 11. Karel Blaha (1926-1988) Appendix 251 Plate 12. Max Brenner Plate 13. Hans Brockmann (1903-1988) Plate 14. Victor Bruckner (1900- 1980) Plate 15. Pehr V. Edman (1916- 1977) 252 Appendix Plate 16. Lyman C. Craig (1906-1974) Plate 17. Vittorio Erspamer Plate 18. Joseph S. Fruton, Biochemist and Historian Appendix 253 Plate 19. Rolf Geiger (1923-1988) Plate 20. Wolfgang Konig Plate 21. Dorothy Hodgkins Plate. 22. Franz Hofmeister (1850-1922), (Fischer, biograph. Lexikon) 254 Appendix Plate 23. The picture shows the late Professor 1.E. Jorpes (r.j and Professor V. Mutt during their favorite pastime in the archipelago on the Baltic near Stockholm Plate 24. Ephraim Katchalski (Katzir) Plate 25. Abraham Patchornik Appendix 255 Plate 26. P.G. Katsoyannis Plate 27. George W. Kenner (1922-1978) Plate 28. Edger Lederer (1908- 1988) Plate 29. Hennann Leuchs (1879-1945) 256 Appendix Plate 30. Choh Hao Li (1913-1987) Plate 31. -

Than Was the Monoleucyl Ester of Cis-Cyclopentane-1,2-Diol
SOME OBSERVATIONS ON THE MECHANISM OF THE ACYLATION PROCESS IN PROTEIN SYNTHESIS* BY BEVERLY E. GRIFFIN AND C. B. REESE UNIVERSITY CHEMICAL LABORATORY, CAMBRIDGE, ENGLAND Communicated by Lord Todd, January 2, 1964 During recent years much attention has been given to the mechanism of synthesis of proteins in biological systems.' Although the process is by no means fully understood, some of the steps involved appear to be well established.2-4 Before amino acids can be incorporated into polypeptide chains they must first be "activated." The "activation" process involves ribonucleic acids of comparatively low molecular weight, known as "transfer" or "soluble" ribonucleic acids (sRNA), adenosine-5' triphosphate (ATP) as an energy source, and a specific enzyme for each amino acid. In the first step of the "activation" process, the amino acid reacts with ATP to form a mixed anhydride with adenosine-5' phosphate (AMP) releasing inorganic pyrophosphate; this mixed anhydride then acylates a specific sRNA on its terminal nucleoside (adenosine) residue to yield a 2' (or 3')-aminoacyl ester-the "activated" amino-acid. These processes are reversible. ATP + amino acid aminoacyl-AMP + pyrophosphate (1) sRNA + aminoacyl-AMP aminoacyl-sRNA + AMP (2) Beyond this point our knowledge of the process of polypeptide synthesis is less certain. The "activated" amino acids are believed to be transferred to the ribo- somes-the site of assembly of polypeptide chains-and then the amino acids become linked together in a genetically controlled order to synthesize a specific protein.5 Although there is as yet no definite evidence regarding this final step, it is frequently assumed that it involves a simple acylation as indicated in step (3). -

Carboligation Using the Aldol Reaction
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1730 Carboligation using the aldol reaction A comparison of stereoselectivity and methods DERAR AL-SMADI ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 ISBN 978-91-513-0472-4 UPPSALA urn:nbn:se:uu:diva-362866 2018 Dissertation presented at Uppsala University to be publicly examined in BMC C2:301, Husargatan 3, Uppsala, Friday, 30 November 2018 at 09:15 for the degree of Doctor of Philosophy. The examination will be conducted in English. Faculty examiner: Professor Ulf Nilsson (Lund University). Abstract Al-Smadi, D. 2018. Carboligation using the aldol reaction. A comparison of stereoselectivity and methods. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1730. 50 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-513-0472-4. The research summarized in this thesis focuses on synthesizing aldehyde and aldol compounds as substrates and products for the enzyme D-fructose-6-aldolase (FSA). Aldolases are important enzymes for the formation of carbon-carbon bonds in nature. In biological systems, aldol reactions, both cleavage and formation play central roles in sugar metabolism. Aldolases exhibit high degrees of stereoselectivity and can steer the product configurations to a given enantiomeric and diastereomeric form. To become truly useful synthetic tools, the substrate scope of these enzymes needs to become broadened. In the first project, phenylacetaldehyde derivatives were synthesized for the use as test substrates for E. coli FSA. Different methods were discussed to prepare phenylacetaldehyde derivatives, the addition of a one carbon unit to benzaldehyde derivatives using a homologation reaction was successful and was proven efficient and non-sensitive to the moisture. -

Aldehydes Can React with Alcohols to Form Hemiacetals
340 14 . Nucleophilic substitution at C=O with loss of carbonyl oxygen You have, in fact, already met some reactions in which the carbonyl oxygen atom can be lost, but you probably didn’t notice at the time. The equilibrium between an aldehyde or ketone and its hydrate (p. 000) is one such reaction. O HO OH H2O + R1 R2 R1 R2 When the hydrate reverts to starting materials, either of its two oxygen atoms must leave: one OPh came from the water and one from the carbonyl group, so 50% of the time the oxygen atom that belonged to the carbonyl group will be lost. Usually, this is of no consequence, but it can be useful. O For example, in 1968 some chemists studying the reactions that take place inside mass spectrometers needed to label the carbonyl oxygen atom of this ketone with the isotope 18 O. 16 18 By stirring the ‘normal’ O compound with a large excess of isotopically labelled water, H 2 O, for a few hours in the presence of a drop of acid they were able to make the required labelled com- í In Chapter 13 we saw this way of pound. Without the acid catalyst, the exchange is very slow. Acid catalysis speeds the reaction up by making a reaction go faster by raising making the carbonyl group more electrophilic so that equilibrium is reached more quickly. The the energy of the starting material. We 18 also saw that the position of an equilibrium is controlled by mass action— O is in large excess. -

3 Alkenes from 1,2-Diols
REVISTA BOLIVIANA DE QUÍMICA (Rev.Bol.Quim.) Vol. 32, No.5, pp. 121-125, Nov./Dic. 2015 Bolivian Journal of Chemistry 32(5) 121-125, Nov./Dec. 2015 Received 12 14 2015 Accepted 12 23 2015 Published 12 30 2015 Bravo et Vila . STEREOSPECIFIC SYNTHESIS OF ALKENES FROM 1,2-DIOLS; MECHANISTIC VIEWS; THE ORGANIC CHEMISTRY NOTEBOOK SERIES, A DIDACTICAL APPROACH, Nº 8 José A. Bravo 1,*, José L. Vila 2 1Department of Chemistry, Laboratorio de Fitoquímica, Instituto de Investigaciones en Productos Naturales IIPN, Universidad Mayor de San Andrés UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota, Phone 59122792238, La Paz, Bolivia, [email protected] 2Department of Chemistry, Laboratorio de Síntesis y Hemisíntesis, Instituto de Investigaciones en Productos Naturales IIPN, Universidad Mayor de San Andrés UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota, Phone 59122795878, La Paz, Bolivia, [email protected] Keywords: Organic Chemistry, Alkenes, 1,2-diols, Stereospecific synthesis, Mechanisms of Reactions, W. Carruthers. ABSTRACT This is the eighth chapter in the series: “The Organic Chemistry Notebook Series, a Didactical Approach”. The aim of this series of studies is to help students to have a graphical view of organic synthesis reactions of diverse nature. Here we discuss, from a mechanistic stand point, some methods for the stereospecific synthesis of alkenes from 1,2-diols. One of the best ones utilizes as precursors, the cyclic thionocarbonates obtained from the diol with thiophosgene. We describe by mechanisms, the use of 1,3-dimethyl-2-phenyl-1,3,2-diazophospholidine as an alternative for the decomposition of thionocarbonates into alkenes. -

Chapter 19 the Chemistry of Aldehydes and Ketones. Addition Reactions
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 19 The Chemistry of Aldehydes and Ketones. Addition Reactions Solutions to In-Text Problems 19.1 (b) (d) (e) (g) 19.2 (a) 2-Propanone (d) (E)-3-Ethoxy-2-propenal (f) 4,4-Dimethyl-2,5-cyclohexadienone 19.3 (b) 2-Cyclohexenone has a lower carbonyl stretching frequency because its two double bonds are conjugated. 19.4 (b) The compound is 2-butanone: (c) The high frequency of the carbonyl absorption suggests a strained ring. (See Eq. 19.4, text p. 897.) In fact, cyclobutanone matches the IR stretching frequency perfectly and the NMR fits as well: 19.6 The structure and CMR assignments of 2-ethylbutanal are shown below. The two methyl groups are chemically equivalent, and the two methylene groups are chemically equivalent; all carbons with different CMR chemical shifts are chemically nonequivalent. INSTRUCTOR SUPPLEMENTAL SOLUTIONS TO PROBLEMS • CHAPTER 19 2 19.7 (a) The double bonds in 2-cyclohexenone are conjugated, but the double bonds in 3-cyclohexenone are not. Consequently, 2-cyclohexenone has the UV spectrum with the greater lmax. 19.9 Compound A, vanillin, should have a p T p* absorption at a greater lmax when dissolved in NaOH solution because the resulting phenolate can delocalize into the carboxaldehyde group; the resulting phenolate from compound B, isovanillin, on the other hand, can only delocalize in the aromatic ring. 19.11 The mass spectrum of 2-heptanone should have major peaks at m/z = 43 (from a-cleavage), 71 (from inductive cleavage), and 58 (from McLafferty rearrangement). -

Lecture 1: Strategies and Tactics in Organic Synthesis
Massachusetts Institute of Technology Organic Chemistry 5.511 October 3, 2007 Prof. Rick L. Danheiser Lecture 1: Strategies and Tactics in Organic Synthesis Retrosynthetic Analysis The key to the design of efficient syntheses ". the grand thing is to be able to reason backwards. That is a very useful accomplishment, and a very easy "The end is where we start from...." one, but people do not practice it much." T. S. Eliot, in "The Four Quartets" Sherlock Holmes, in "A Study in Scarlet" Strategy Tactics overall plan to achieve the means by which plan ultimate synthetic target is implemented intellectual experimental retrosynthetic planning synthetic execution TRANSFORMS REACTIONS Target Precursor Precursor Target Definitions Retron Structural unit that signals the application of a particular strategy algorithm during retrosynthetic analysis. Transform Imaginary retrosynthetic operation transforming a target molecule into a precursor molecule in a manner such that bond(s) can be reformed (or cleaved) by known or reasonable synthetic reactions. Strategy Algorithm Step-by-step instructions for performing a retrosynthetic operation. "...even in the earliest stages of the process of simplification of a synthetic problem, the chemist must make use of a particular form of analysis which depends on the interplay between structural features that exist in the target molecule and the types of reactions or synthetic operations available from organic chemistry for the modification or assemblage of structural units. The synthetic chemist has learned by experience to recognize within a target molecule certain units which can be synthesized, modified, or joined by known or conceivable synthetic operations...it is convenient to have a term for such units; the term "synthon" is suggested. -
![[2.2]Paracyclophanes- Structure and Reactivity Studies Von Der](https://docslib.b-cdn.net/cover/2287/2-2-paracyclophanes-structure-and-reactivity-studies-von-der-1602287.webp)
[2.2]Paracyclophanes- Structure and Reactivity Studies Von Der
Syntheses of Functionalised [2.2]Paracyclophanes- Structure and Reactivity Studies Von der Gemeinsamen Naturwissenschaftlichen Fakultät der Technischen Universität Carolo Wilhelmina zu Braunschweig zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr.rer.nat.) genehmigte D i s s e r t a t i o n von Swaminathan Vijay Narayanan aus Chennai (Madras) / India 1. Referent: Prof. Dr. Dr. h. c. Henning Hopf 2. Referentin: Prof. Dr. Monika Mazik eingereicht am: 20. Jan 2005 mündliche Prüfung (Disputation) am: 29. März 2005 Druckjahr 2005 Vorveröffentlichungen der Dissertation Teilergebnisse aus dieser Arbeit wurden mit Genehmigung der Gemeinsamen Naturwissenschaft-lichen Fakultät, vertreten durch die Mentorin oder den Mentor/die Betreuerin oder den Betreuer der Arbeit, in folgenden Beiträgen vorab veröffentlicht: Publikationen K. El Shaieb, V. Narayanan, H. Hopf, I. Dix, A. Fischer, P. G. Jones, L. Ernst & K. Ibrom.: 4,15-Diamino[2.2]paracyclophane as a starting material for pseudo-geminally substituted [2.2]paracyclophanes. Eur. J. Org. Chem.: 567-577 (2003). Die vorliegende Arbeit wurde in der Zeit von Oktober 2001 bis Januar 2004 am Institut für Organische Chemie der Technischen Universität Braunschweig unter der Leitung von Prof. Dr. Dr. h.c. Henning Hopf angefertigt. It is my great pleasure to express my sincere gratitude to Prof. Dr. Henning Hopf for his support, encouragement and guidance throughout this research work. I thank him a lot for his invaluable ideas and remarks which made this study very interesting. I admire deep from my heart his energetic way of working and his brilliant ideas which made possible this research work to cover a vast area. -

1,2-Hexanediol
Supporting Information for Low-Priority Substance 1,2- Hexanediol (CASRN 6920-22-5) Final Designation February 20, 2020 Office of Pollution Prevention and Toxics U.S. Environmental Protection Agency 1200 Pennsylvania Avenue Washington, DC 20460 Contents 1. Introduction ................................................................................................................................................................ 1 2. Background on 1,2-Hexanediol ................................................................................................................................. 3 3. Physical-Chemical Properties ................................................................................................................................... 4 3.1 References ....................................................................................................................................................... 6 4. Relevant Assessment History ................................................................................................................................... 7 5. Conditions of Use ....................................................................................................................................................... 8 6. Hazard Characterization .......................................................................................................................................... 10 6.1 Human Health Hazard ................................................................................................................................... -
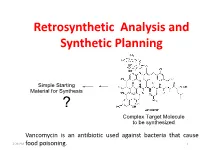
Retrosynthetic Analysis and Synthetic Planning
Retrosynthetic Analysis and Synthetic Planning Vancomycin is an antibiotic used against bacteria that cause 2:24 PM food poisoning. 1 Life’s Perspectives Planning a Journey to the Unknown 2:24 PM 2 Retrosynthetic Analysis Definition Retrosynthetic analysis (retrosynthesis) is a technique for planning a synthesis, especially of complex organic molecules, whereby the complex target molecule (TM) is reduced into a sequence of progressively simpler structures along a pathway which ultimately leads to the identification of a simple or commercially available starting material (SM) from which a chemical synthesis can then be developed. Retrosynthetic analysis is based on known reactions (e.g the Wittig reaction, oxidation, reduction etc). The synthetic plan generated from the retrosynthetic analysis will be the roadmap to guide the synthesis of the target molecule. 2:24 PM 3 Synthetic Planning Definition Synthesis is a construction process that involves converting simple or commercially available molecules into complex molecules using specific reagents associated with known reactions in the retrosynthetic scheme. Syntheses can be grouped into two broad categories: (i) Linear syntheses 2:24 PM (ii)Convergent syntheses 4 Linear Synthesis Definition In linear synthesis, the target molecule is synthesized through a series of linear transformations. Since the overall yield of the synthesis is based on the single longest route to the target molecule, by being long, a linear synthesis suffers a lower overall yield. The linear synthesis is fraught with failure for its lack of flexibility leading to potential large losses in the material already invested in the synthesis at the time of failure. 2:24 PM 5 Convergent Synthesis Definition In convergent synthesis, key fragments of the target molecule are synthesized separately or independently and then brought together at a later stage in the synthesis to make the target molecule.