Pilocytic Astrocytoma of the Spinal Cord
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Leptomeningeal Dissemination of Pilocytic Astrocytoma Via Hematoma in a Child
Neurosurg Focus 13 (1):Clinical Pearl 2, 2002, Click here to return to Table of Contents Leptomeningeal dissemination of pilocytic astrocytoma via hematoma in a child Case report MASARU KANDA, M.D., HIDENOBU TANAKA, M.D., PH.D., SOJI SHINODA, M.D., PH.D., AND TOSHIO MASUZAWA, M.D., PH.D. Department of Surgical Neurology, Jichi Medical School, Tochigi, Japan A case of recurrent pilocytic astrocytoma with leptomeningeal dissemination (LMD) is described. A cerebellar tumor was diagnosed in a 3-year-old boy, in whom resection was performed. When the boy was 6 years of age, recur- rence was treated with surgery and local radiotherapy. At age 13 years, scoliosis was present, but the patient was asymptomatic. Twelve years after initial surgery LMD was demonstrated in the lumbar spinal region without recur- rence of the original tumor. This tumor also was subtotally removed. During the procedure, a hematoma was observed adjacent to the tumor, but the border was clear. Histological examination of the spinal cord tumor showed features sim- ilar to those of the original tumor. There were no tumor cells in the hematoma. The MIB-1 labeling index indicated no malignant change compared with the previous samples. Radiotherapy was performed after the surgery. The importance of early diagnosis and management of scoliosis is emphasized, and the peculiar pattern of dissemination of the pilo- cytic astrocytoma and its treatment are reviewed. KEY WORDS • pilocytic astrocytoma • leptomeningeal dissemination • MIB-1 labeling index • radiation therapy • scoliosis -

Central Nervous System
PRINCESS MARGARET CANCER CENTRE CLINICAL PRACTICE GUIDELINES CENTRAL NERVOUS SYSTEM LOW GRADE GLIOMAS CNS Site Group – Low Grade Gliomas Author: Dr. Norm Laperriere 1. INTRODUCTION 3 2. PREVENTION 3 3. SCREENING AND EARLY DETECTION 3 4. DIAGNOSIS AND PATHOLOGY 3 5. MANAGEMENT 4 5.1 MANAGEMENT ALGORITHMS 4 5.2 SURGERY 4 5.3 CHEMOTHERAPY 5 5.4 RADIATION THERAPY 5 6. ONCOLOGY NURSING PRACTICE 6 7. SUPPORTIVE CARE 6 7.1 PATIENT EDUCATION 6 7.2 PSYCHOSOCIAL CARE 6 7.3 SYMPTOM MANAGEMENT 6 7.4 CLINICAL NUTRITION 7 7.5 PALLIATIVE CARE 7 7.6 REHABILITATION 7 8. FOLLOW-UP CARE 7 Last Revision Date – April 2019 2 Low Grade Gliomas 1. Introduction • Grade I gliomas: pilocytic astrocytoma (PA), dysembryoplastic neuroepithelial tumor (DNET), pleomorphic xanthoastrocytoma, (PXA), ganglioglioma • Grade II gliomas: infiltrating astrocytoma, oligodendroglioma, mixed gliomas • annual incidence is approx. 1/100,000 This document is intended for use by members of the Central Nervous System site group of the Princess Margaret Hospital/University Health Network. The guidelines in this document are meant as a guide only, and are not meant to be prescriptive. There exists a multitude of individual factors, prognostic factors and peculiarities in any individual case, and for that reason the ultimate decision as to the management of any individual patient is at the discretion of the staff physician in charge of that particular patient’s care. 2. Prevention • genetic counseling for all NF1 carriers 3. Screening and Early Detection • baseline MRI brain for all newly -

Central Nervous System Tumors General ~1% of Tumors in Adults, but ~25% of Malignancies in Children (Only 2Nd to Leukemia)
Last updated: 3/4/2021 Prepared by Kurt Schaberg Central Nervous System Tumors General ~1% of tumors in adults, but ~25% of malignancies in children (only 2nd to leukemia). Significant increase in incidence in primary brain tumors in elderly. Metastases to the brain far outnumber primary CNS tumors→ multiple cerebral tumors. One can develop a very good DDX by just location, age, and imaging. Differential Diagnosis by clinical information: Location Pediatric/Young Adult Older Adult Cerebral/ Ganglioglioma, DNET, PXA, Glioblastoma Multiforme (GBM) Supratentorial Ependymoma, AT/RT Infiltrating Astrocytoma (grades II-III), CNS Embryonal Neoplasms Oligodendroglioma, Metastases, Lymphoma, Infection Cerebellar/ PA, Medulloblastoma, Ependymoma, Metastases, Hemangioblastoma, Infratentorial/ Choroid plexus papilloma, AT/RT Choroid plexus papilloma, Subependymoma Fourth ventricle Brainstem PA, DMG Astrocytoma, Glioblastoma, DMG, Metastases Spinal cord Ependymoma, PA, DMG, MPE, Drop Ependymoma, Astrocytoma, DMG, MPE (filum), (intramedullary) metastases Paraganglioma (filum), Spinal cord Meningioma, Schwannoma, Schwannoma, Meningioma, (extramedullary) Metastases, Melanocytoma/melanoma Melanocytoma/melanoma, MPNST Spinal cord Bone tumor, Meningioma, Abscess, Herniated disk, Lymphoma, Abscess, (extradural) Vascular malformation, Metastases, Extra-axial/Dural/ Leukemia/lymphoma, Ewing Sarcoma, Meningioma, SFT, Metastases, Lymphoma, Leptomeningeal Rhabdomyosarcoma, Disseminated medulloblastoma, DLGNT, Sellar/infundibular Pituitary adenoma, Pituitary adenoma, -

Differentiation Between Pilocytic Astrocytoma and Glioblastoma: a Decision Tree Model Using Contrast-Enhanced Magnetic Resonance
European Radiology (2019) 29:3968–3975 https://doi.org/10.1007/s00330-018-5706-6 ONCOLOGY Differentiation between pilocytic astrocytoma and glioblastoma: a decision tree model using contrast-enhanced magnetic resonance imaging-derived quantitative radiomic features Fei Dong1 & Qian Li1 & Duo Xu1 & Wenji Xiu2 & Qiang Zeng3 & Xiuliang Zhu1 & Fangfang Xu1 & Biao Jiang1 & Minming Zhang1 Received: 13 March 2018 /Revised: 8 July 2018 /Accepted: 6 August 2018 /Published online: 12 November 2018 # European Society of Radiology 2018 Abstract Objective To differentiate brain pilocytic astrocytoma (PA) from glioblastoma (GBM) using contrast-enhanced mag- netic resonance imaging (MRI) quantitative radiomic features by a decision tree model. Methods Sixty-six patients from two centres (PA, n = 31; GBM, n = 35) were randomly divided into training and validation data sets (about 2:1). Quantitative radiomic features of the tumours were extracted from contrast-enhanced MR images. A subset of features was selected by feature stability and Boruta algorithm. The selected features were used to build a decision tree model. Predictive accuracy, sensitivity and specificity were used to assess model performance. The classification outcome of the model was combined with tumour location, age and gender features, and multivariable logistic regression analysis and permutation test using the entire data set were performed to further evaluate the decision tree model. Results A total of 271 radiomic features were successfully extracted for each tumour. Twelve features were selected as input variables to build the decision tree model. Two features S(1, -1) Entropy and S(2, -2) SumAverg were finally included in the model. The model showed an accuracy, sensitivity and specificity of 0.87, 0.90 and 0.83 for the training data set and 0.86, 0.80 and 0.91 for the validation data set. -

Malignant CNS Solid Tumor Rules
Malignant CNS and Peripheral Nerves Equivalent Terms and Definitions C470-C479, C700, C701, C709, C710-C719, C720-C725, C728, C729, C751-C753 (Excludes lymphoma and leukemia M9590 – M9992 and Kaposi sarcoma M9140) Introduction Note 1: This section includes the following primary sites: Peripheral nerves C470-C479; cerebral meninges C700; spinal meninges C701; meninges NOS C709; brain C710-C719; spinal cord C720; cauda equina C721; olfactory nerve C722; optic nerve C723; acoustic nerve C724; cranial nerve NOS C725; overlapping lesion of brain and central nervous system C728; nervous system NOS C729; pituitary gland C751; craniopharyngeal duct C752; pineal gland C753. Note 2: Non-malignant intracranial and CNS tumors have a separate set of rules. Note 3: 2007 MPH Rules and 2018 Solid Tumor Rules are used based on date of diagnosis. • Tumors diagnosed 01/01/2007 through 12/31/2017: Use 2007 MPH Rules • Tumors diagnosed 01/01/2018 and later: Use 2018 Solid Tumor Rules • The original tumor diagnosed before 1/1/2018 and a subsequent tumor diagnosed 1/1/2018 or later in the same primary site: Use the 2018 Solid Tumor Rules. Note 4: There must be a histologic, cytologic, radiographic, or clinical diagnosis of a malignant neoplasm /3. Note 5: Tumors from a number of primary sites metastasize to the brain. Do not use these rules for tumors described as metastases; report metastatic tumors using the rules for that primary site. Note 6: Pilocytic astrocytoma/juvenile pilocytic astrocytoma is reportable in North America as a malignant neoplasm 9421/3. • See the Non-malignant CNS Rules when the primary site is optic nerve and the diagnosis is either optic glioma or pilocytic astrocytoma. -

Perinatal (Fetal and Neonatal) Astrocytoma: a Review
Childs Nerv Syst DOI 10.1007/s00381-016-3215-y REVIEW PAPER Perinatal (fetal and neonatal) astrocytoma: a review Hart Isaacs Jr.1,2 Received: 16 July 2016 /Accepted: 3 August 2016 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Keywords Fetal astrocytoma . Neonatal astrocytoma . Introduction The purpose of this review is to document the Perinatal astrocytoma . Intracranial hemorrhage . Congenital various types of astrocytoma that occur in the fetus and neo- brain tumor nate, their locations, initial findings, pathology, and outcome. Data are presented that show which patients are likely to sur- vive or benefit from treatment compared with those who are Introduction unlikely to respond. Materials and methods One hundred one fetal and neonatal Glial cells are the supportive elements of the central nervous tumors were collected from the literature for study. system (CNS) [22]. They include astrocytes, oligodendro- Results Macrocephaly and an intracranial mass were the most cytes, and ependymal cells, and the corresponding tumors common initial findings. Overall, hydrocephalus and intracra- originating from these cells astrocytoma, oligodendroglioma, nial hemorrhage were next. Glioblastoma (GBM) was the and ependymoma all of which are loosely called Bglioma^ most common neoplasm followed in order by subependymal [16, 22]. The term Bglioma^ is used interchangeably with giant cell astrocytoma (SEGA), low-grade astrocytoma, ana- astrocytoma to describe the more common subgroup of tu- plastic astrocytoma, and desmoplastic infantile astrocytoma mors [22]. (DIA). Tumors were detected most often toward the end of Glioma (astrocytoma) is the leading CNS tumor in chil- the third trimester of pregnancy. -

Neuropathology Neoadjuvant Chemoradiation and Pancreatectomy
378A ANNUAL MEETING ABSTRACTS 1602 Pathologic Complete Response Is Associated with Good Prognosis in Patient with Pancreatic Ductal Adenocarcinoma Who Received Neuropathology Neoadjuvant Chemoradiation and Pancreatectomy. Q Zhao, A Rashid, Y Gong, M Katz, J Lee, R Wolf, C Charnsangavej, G Varadhachary, P 1604 Nestin, an Important Marker for Differentiating Oligodendroglioma Pisters, E Abdalla, J-N Vauthey, H Wang, H Gomez, J Fleming, J Abbruzzese, H Wang. from Astrocytic Tumors. MD Anderson Cancer Center, Houston, TX. SH Abu-Farsakh, IA Sbeih, HA Abu-Farsakh. Jordan University, Amman, Jordan; Ibn Background: Patients with pancreatic ductal adenocarcinoma (PDA) has poor Hytham Hospital, Amman, Jordan; First Medical Lab, Amman, Jordan. prognosis. To improve the clinical outcome, most patients with PDA are treated with Background: Nestin is an acronym for neuroepithelial stem cell protein. It is an neoadjuvant chemoradiation prior to surgery at our institution. In this group of patients, intermediate filament protein expressed in proliferating cells during the developmental pathologic complete response (PCR) is rarely observed in subsequent pancreatectomies. stages in a variety of embryonic and fetal tissues. It is also expressed in some adult stem/ However, the prognostic significance of PCR is not clear. progenitor cell populations, such as newborn vascular endothelial cell. Differentiation Design: Among 442 patients with PDA who received neoadjuvant chemoradiation and between astrocytic tumors and oligodenoglioma tumor is of paramount importance pancreatectomy from 1995 to 2010, 11 (2%) patients with PCR were identified. The because of different lines of treatment and different prognosis. cytologic diagnosis on pre-therapy tumor was reviewed and PCR in pancreatectomies Design: We performed Nestin immunostaining on paraffin blocks of 16 cases of was confirmed in all patients. -
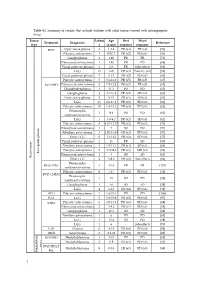
Table S1. Summary of Studies That Include Children with Solid Tumors Treated with Antiangiogenic Drugs
Table S1. Summary of studies that include children with solid tumors treated with antiangiogenic drugs. Tumor Patient Age Best Worst Treatment Diagnostic Reference type s (years) response response BVZ Optic nerve glioma 4 1.2-4 PR (x4) PR (x4) [56] Pilocytic astrocytoma 3 0.92-7 PR (x2) PD (x1) [56] Ganglioglioma 1 1.92 PR PR [56] Pilomyxoid astrocytoma 1 1.92 PR PD [68] Visual pathway glioma 1 2.6 PR Side effects [66] LGG 15 1-20 CR (x3) Toxicity (x1) [58] Visual pathway glioma 3 6-13 PR (x2) PD (x1) [60] Pilocytic astrocytoma 5 3.1-11.2 PR (x5) PR (x5) [65] BVZ+IRO Pilomyxoid astrocytoma 2 7.9-12.2 PR (x2) PR (x2) [65] Oligodendroglioma 1 11.1 PD PD [65] Ganglioglioma 3 4.1-16.8 PR (x2) SD (x1) [65] Optic nerve glioma 2 9-15 PR (x1) SD (x1) [63] LGG 35 0.6-17.6 PR (x2) PD (x8) [61] Pilocytic astrocytoma 10 1.8-15.3 PR (x4) PD (x1) [62] Pleomorphic 1 9.4 PD PD [62] xanthoastrocytoma LGG 5 3.9-9.2 PR (x2) SD (x3) [62] Pilocytic astrocytoma 4 4.67-12.17 PR (x2) PD (x3) [57] Pilomyxoid astrocytoma 1 3 SD PD [57] Fibrillary astrocytoma 3 1.92-11.08 CR (x1) PD (x3) [57] Other LGG 6 1-13.42 PR (x2) PD (x6) [57] Visual pathway glioma 1 11 PR SD [60] Fibrillary astrocytoma 3 1.5-11.1 CR (x1) SD (x1) [64] Pilocytic astrocytoma 2 3.75-9.8 PR (x1) MR (x1) [64] Low-grade glioma Pilomyxoid astrocytoma 1 3 SD SD [64] Brain tumor Brain tumor Other LGG 4 3-9.6 PR (x2) Side effects [64] Pleomorphic BVZ+TMZ 1 13.4 PR PR [153] xanthoastrocytoma Pilocytic astrocytoma 9 5-17 PR (x3) PD (x3) [59] BVZ+CHEM Pleomorphic 1 18 SD PD [59] xanthoastrocytoma Ganglioglioma -

Recurrent Cerebral Astroblastoma with Aggressive Histology: an Uncommon Case
THIEME 150 Case Report Recurrent Cerebral Astroblastoma with Aggressive Histology: An Uncommon Case Sumit Bansal1 Susama Patra2 Pritinanda Mishra2 Ashok Kumar Mahapatra1 1 Department of Neurosurgery, All India Institute of Medical Address for correspondence Dr. Sumit Bansal, MCh, Room No. 419, Sciences, Bhubaneswar, Odisha, India Department of Neurosurgery, All India Institute of Medical Sciences, 2 Department of Pathology and Laboratory Medicine, All India Bhubaneswar, Odisha 751019, India Institute of Medical Sciences, Bhubaneswar, Odisha, India (e-mail: [email protected]). Indian J Neurosurg 2018;7:150–154 Abstract Astroblastomas are uncommon tumors of uncertain histogenesis. Recently tanycyte is postulated to be the cell of origin. These occur predominantly in the cerebral hemisphere of young adults and children. Astroblastoma constitutes approximately 0.45 to 2.8% of all neuroglial tumors. We are reporting a case of 28-year-old woman who present- ed with headache and double vision. Magnetic resonance imaging (MRI) of the brain revealed well-demarcated, ring-enhancing solid cystic mass in right temporoparietal lobe with significant mass effect. The patient underwent gross total resection of the lesion through right temporoparietal craniotomy. The histopathologic diagnosis was suggestive of astroblastoma. She was advised for postoperative radiotherapy, but she lost to follow-up and again presented 1 year later with recurrent tumor in the same Keywords location along with another lesion in infratentorial location. She was reoperated and ► astroblastoma gross total excision of supra- and infratentorial tumor was done. She also received ► malignant radiotherapy. We are reporting this uncommon case of astroblastoma and also review- ► cystic ing the existing literature. Introduction Case Report Astroblastomas are unusual tumors, constituting 0.45 to 2.8% A 28-year-old woman presented to us with holocranial head- of all neuroglial tumors.1 The cell of origin of astroblastoma ache and double vision for 2 months. -

Current Understanding of Neurofibromatosis Type 1, 2, And
International Journal of Molecular Sciences Review Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis Ryota Tamura Department of Neurosurgery, Kawasaki Municipal Hospital, Shinkawadori, Kanagawa, Kawasaki-ku 210-0013, Japan; [email protected] Abstract: Neurofibromatosis (NF) is a neurocutaneous syndrome characterized by the development of tumors of the central or peripheral nervous system including the brain, spinal cord, organs, skin, and bones. There are three types of NF: NF1 accounting for 96% of all cases, NF2 in 3%, and schwannomatosis (SWN) in <1%. The NF1 gene is located on chromosome 17q11.2, which encodes for a tumor suppressor protein, neurofibromin, that functions as a negative regulator of Ras/MAPK and PI3K/mTOR signaling pathways. The NF2 gene is identified on chromosome 22q12, which encodes for merlin, a tumor suppressor protein related to ezrin-radixin-moesin that modulates the activity of PI3K/AKT, Raf/MEK/ERK, and mTOR signaling pathways. In contrast, molecular insights on the different forms of SWN remain unclear. Inactivating mutations in the tumor suppressor genes SMARCB1 and LZTR1 are considered responsible for a majority of cases. Recently, treatment strategies to target specific genetic or molecular events involved in their tumorigenesis are developed. This study discusses molecular pathways and related targeted therapies for NF1, NF2, and SWN and reviews recent clinical trials which involve NF patients. Keywords: neurofibromatosis type 1; neurofibromatosis type 2; schwannomatosis; molecular tar- geted therapy; clinical trial Citation: Tamura, R. Current Understanding of Neurofibromatosis Int. Type 1, 2, and Schwannomatosis. 1. Introduction J. Mol. Sci. 2021, 22, 5850. https:// doi.org/10.3390/ijms22115850 Neurofibromatosis (NF) is a genetic disorder that causes multiple tumors on nerve tissues, including brain, spinal cord, and peripheral nerves [1–3]. -

Low-Grade Gliomas 26
C H A P T E R Low-Grade Gliomas 26 Paul D. Brown, Michael D. Chan, Edward G. Shaw, and Martin J. van den Bent EPIDEMIOLOGIC AND ETIOLOGIC FINDINGS PRIMARY THERAPY The estimated annual incidence of low-grade gliomas in the Maximum surgical resection is associated with a more favor- United States is 2000 cases. Etiologic factors are unknown. able outcome. These tumors are seen with increased frequency in patients In pediatric low-grade gliomas, observation is recom- with types 1 and 2 neurofibromatosis. mended after gross total resection. After subtotal resection or biopsy, treatment (radiation therapy, chemotherapy) is usually PATHOLOGIC FINDINGS deferred until symptomatic or imaging progression. The most common pathologic types include pilocytic astrocy- In adults, after gross total resection of a pilocytic astrocy- toma (World Health Organization [WHO] grade I), diffuse toma, observation is recommended. After gross total or subto- fibrillary astrocytoma, oligoastrocytoma, and oligodendrogli- tal resection (or biopsy) of a low-grade glioma, irradiation oma (WHO grade II). may be given postoperatively or deferred until progression occurs. BIOLOGIC CHARACTERISTICS The recommended radiotherapy (RT) dose is 45 Gy to Combined 1p and 19q deletions are associated with a superior 54 Gy in 25 fractions to 30 fractions, treating the MRI-defined outcome and are most common in oligodendrogliomas. TP53 tumor volume with a 2-cm margin. mutations are more common in diffuse astrocytomas and are Early results from one large randomized clinical trial mutually exclusive from 1p/19q co-deletions. IDH1 mutations suggest disease and outcomes are similar for either temozolo- occur in the vast majority of low-grade gliomas and are found mide or RT for 1p/19q-deleted tumors although RT may have both in tumors with TP53 mutations and in tumors with a superior PFS for 1p-intact tumors; however, the period of 1p/19q codeletions. -

Impact of RNA-Sequencing Analysis in Refining Diagnosis in Pediatric Neuro-Oncology Kathleen M
Impact of RNA-sequencing analysis in refining diagnosis in pediatric neuro-oncology Kathleen M. Schieffer1, Katherine E. Miller1, Stephanie LaHaye1, James Fitch1, Kyle Voytovich1, Daniel C. Koboldt1, Benjamin Kelly1, Patrick Brennan1, Anthony Miller1, Elizabeth Varga1, Kristen Leraas1, Vibhuti Agarwal2, Diana Osorio2, Mohamed S. AbdelBaki2, Jonathan L. Finlay2, Jeffrey R. Leonard3, Alex Feldman4, Daniel R. Boue4, Julie M. Gastier-Foster1, Vincent Magrini1, Peter White1, Catherine E. Cottrell1, Elaine R. Mardis1, Richard K. Wilson1 1. Institute for Genomic Medicine, Nationwide Children’s Hospital, Columbus, OH; 2. Department of Pediatrics, Nationwide Children’s Hospital, Columbus, OH; 3. Department of Neurosurgery, Nationwide Children’s Hospital, Columbus, OH; 4. Department of Pathology and Laboratory Medicine, Nationwide Children’s Hospital, Columbus, OH Background and demographics Methods Case 2: Refine diagnosis The Nationwide Children’s Hospital Institute for Genomic Medicine (IGM) translational research protocol Transcriptomic analysis was performed from 70 tissue sections collected from 56 patients: snap frozen (n=48), A 10-month-old was diagnosed with a sella/suprasellar tumor, most consistent with a CNS embryonal tumor. No enrolls pediatric patients with high-risk, relapsed, refractory, or difficult to classify tumors. The goal of formalin-fixed paraffin-embedded (FFPE) tissue (n=21), or disassociated cells (n=1). When possible, total RNA- constitutional or somatic variations or copy number aberrations were noted. The tissue was also sent to an this protocol is to refine the patient diagnosis, identify targeted therapeutics relevant to the individual sequencing was performed (n=60); however, when the sample was poor quality or low input, cDNA capture was outside institution for the Infinium MethylationEPIC 850K array, reporting a classification of pineoblastoma group tumor biology and eliminate unsuitable treatments, and determine eligibility for clinical trials.