Manipulating Ig Production in Vivo Through CD180 Stimulation Jay
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

(RP105) Activates B Cells to Rapidly Produce Polyclonal Ig Via a T Cell and Myd88-Independent Pathway
Anti-CD180 (RP105) Activates B Cells To Rapidly Produce Polyclonal Ig via a T Cell and MyD88-Independent Pathway This information is current as Jay W. Chaplin, Shinji Kasahara, Edward A. Clark and of October 6, 2021. Jeffrey A. Ledbetter J Immunol 2011; 187:4199-4209; Prepublished online 14 September 2011; doi: 10.4049/jimmunol.1100198 http://www.jimmunol.org/content/187/8/4199 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2011/09/14/jimmunol.110019 Material 8.DC1 http://www.jimmunol.org/ References This article cites 33 articles, 16 of which you can access for free at: http://www.jimmunol.org/content/187/8/4199.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on October 6, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2011 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Anti-CD180 (RP105) Activates B Cells To Rapidly Produce Polyclonal Ig via a T Cell and MyD88-Independent Pathway Jay W. -

Supplemental Figures 032819.Pptx
Summary of Supplemental Figures and Tables 1) Supplemental Figure 1. Frequency of phenotypically defined endothelial cells is unchanged in aged mice. 2) Supplemental Figure 2. Aged LSKs have increased myeloid/megakaryocytic bias 3) Supplemental Figure 3. RBCs are decreased and Platelets are increased in peripheral blood of aged mice 4) Supplemental Figure 4. Aged BMME cultures have increased MSC populations. 5) Supplemental Figure 5. Aged BMME cells increase young LSK cell engraftment following competitive transplantation 6) Supplemental Figure 6. Flow cytometry gating strategy for sorting of young and aged murine Mφs 7) Supplemental Figure 7. Flow cytometry gating strategy for sorting of human Mφs 8) Supplemental Figure 8. Axl-/- LT-HSCs have increased cell engraftment following competitive transplantation 9) Supplemental Figure 9. Schematic representation of mechanisms by which age-dependent defects in marrow Mφs induce megakaryocytic bias in HSC. 10) Table S1. List of antibodies used in the flow cytometric and cell sorting analyses described 11) Table S2. List of cell populations analyzed and their respective immunophenotypes 12) Table S3. List of top GO-BP categories enriched for significantly upregulated genes from murine aged vs young marrow macrophages 13) Table S4. List of significant KEGG Pathways enriched for significantly upregulated genes from murine aged vs young marrow macrophages 1 A Sinusoidal Arteriolar BV786 – CD31 Sca1 – PE-Cy7 B C Arteriolar EC Sinusoidal ECs (Lin-, CD45-, CD31+ Sca1+) (CD45-Lin-CD31+Sca1-) 0.08 0.15 0.06 0.10 0.04 0.05 0.02 PercentLiveof Cells PercentLiveof Cells 0.00 0.00 Young Aged Young Aged Supplemental Figure 1. -

SUPPLEMENTARY METHODS Cell Culture.-Human Peripheral Blood
SUPPLEMENTARY METHODS Cell culture.-Human peripheral blood mononuclear cells (PBMC) were isolated from buffy coats from normal donors over a Lymphoprep (Nycomed Pharma) gradient. Monocytes were purified from PBMC by magnetic cell sorting using CD14 microbeads (Miltenyi Biotech). Monocytes were cultured at 0.5 x 106 cells/ml for 7 days in RPMI 1640 (standard RPMI, which contains 1 mg/L folic acid) supplemented with 10% fetal calf serum, at 37ºC in a humidified atmosphere with 5% CO2, and containing GM-CSF (1000U/ml) or M-CSF (10 ng/ml, ImmunoTools) to generate GM-CSF- polarized macrophages (GM-MØ) or M-CSF-polarized macrophages (M-MØ). MTX pharmacokinetic studies in RA patients administered 25 mg MTX showed peak plasma levels of 1-2 µM MTX two hours after drug administration, but plasma levels decline to 10-50 nM MTX within 24-48 hours [1, 2]. MTX (50 nM), pemetrexed (PMX, 50 nM), folic acid (FA, 50 nM) [3], thymidine (dT, 10 µM), pifithrin-α (PFT, 25-50 µM), nutlin-3 (10 µM, Sigma-Aldrich) was added once on monocytes together with the indicated cytokine, or on monocytes and 7-day differentiated macrophages for 48h. Gene expression profiling.-For long-term MTX treatment, RNA was isolated from three independent preparations of monocytes either unexposed or exposed to MTX (50 nM) and differentiated to GM-MØ or M-MØ for 7-days. For short-term schedule, RNA was isolated from three independent samples of fully differentiated GM-MØ either unexposed or exposed to MTX (50 nM) for 48h, by using RNeasy Mini kit (QIAGEN). -
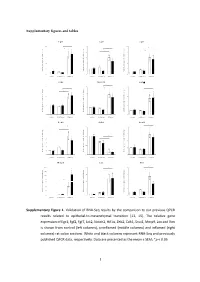
Type of the Paper (Article
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

Treatment Strategies Considering Micro-Environment and Clonal Evolution in Multiple Myeloma
cancers Review Treatment Strategies Considering Micro-Environment and Clonal Evolution in Multiple Myeloma Kazuhito Suzuki 1,2,*, Kaichi Nishiwaki 1,2 and Shingo Yano 2 1 Division of Clinical Oncology/Hematology, Department of Internal Medicine, The Jikei University Kashiwa Hospital, Kashiwa-shita 163-1, Kashiwa-city, Chiba 277-8567, Japan; [email protected] 2 Division of Clinical Oncology/Hematology, Department of Internal Medicine, The Jikei University School of Medicine, Nishi-shimbashi 3-25-1, Minato-ku, Tokyo 105-8461, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-471-64-1111 Simple Summary: Multiple myeloma is an uncurable hematological malignancy, although the prognosis of myeloma patients is getting better using proteasome inhibitors (PIs), immune modula- tory drugs (IMiDs), monoclonal antibodies (MoAbs), and cytotoxic agents. Drug resistance makes myeloma difficult to treat and it can be subdivided into two broad categories: de novo and acquired. De novo drug resistance is associated with the bone marrow microenvironment including bone mar- row stromal cells, the vascular niche and endosteal niche. Acquired drug resistance is related to clonal evolution and non-genetic diversity. The initial treatment plays the most important role considering de novo and acquired drug resistance and should contain PIs, IMIDs, MoAbs, and autologous stem cell transplantation because these treatments improve the bone marrow microenvironment and might prevent clonal evolution via sustained deep response including minimal residual disease negativity. Abstract: Multiple myeloma is an uncurable hematological malignancy because of obtained drug resistance. Microenvironment and clonal evolution induce myeloma cells to develop de novo and acquired drug resistance, respectively. -

Cell-Surface Expression of the TLR Homolog CD180 in Circulating Cells from Splenic and Nodal Marginal Zone Lymphomas
Letters to the Editor 1748 REFERENCES fusion of AF10 and MLL is resolved by fluorescent in situ hybridization analysis. 1 Chaplin T, Ayton P, Bernard OA, Saha V, Della Valle V, Hillion J et al. A novel class of Cancer Res 1995; 55: 4220–4224. zinc finger/leucine zipper genes identified from the molecular cloning of the 6 DiMartino JF, Ayton PM, Chen EH, Naftzger CC, Young BD, Cleary ML. The AF10 t(10;11) translocation in acute leukemia. Blood 1995; 85: 1435–1441. leucine zipper is required for leukemic transformation of myeloid progenitors by 2 Chaplin T, Bernard O, Beverloo HB, Saha V, Hagemeijer A, Berger R et al. MLL-AF10. Blood 2002; 99: 3780–3785. The t(10;11) translocation in acute myeloid leukemia (M5) consistently fuses the 7 Van Limbergen H, Poppe B, Janssens A, De Bock R, De Paepe A, Noens L et al. leucine zipper motif of AF10 onto the HRX gene. Blood 1995; 86: 2073–2076. Molecular cytogenetic analysis of 10;11 rearrangements in acute myeloid leukemia. 3 Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J et al. New insights to Leukemia 2002; 16: 344–351. the MLL recombinome of acute leukemias. Leukemia 2009; 23: 1490–1499. 8 Chen J, Feng W, Jiang J, Deng Y, Huen MS. Ring finger protein RNF169 antagonises 4 Morerio C, Rapella A, Tassano E, Rosanda C, Panarello C. MLL-MLLT10 the ubiquitin-dependent signaling cascade at sites of dna damage. J Biol Chem fusion gene in pediatric acute megakaryoblastic leukemia. Leuk Res 2005; 29: 2012; 287: 27715–27722. -

Human CD Marker Chart Reviewed by HLDA1 Bdbiosciences.Com/Cdmarkers
BD Biosciences Human CD Marker Chart Reviewed by HLDA1 bdbiosciences.com/cdmarkers 23-12399-01 CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD1a R4, T6, Leu6, HTA1 b-2-Microglobulin, CD74 + + + – + – – – CD93 C1QR1,C1qRP, MXRA4, C1qR(P), Dj737e23.1, GR11 – – – – – + + – – + – CD220 Insulin receptor (INSR), IR Insulin, IGF-2 + + + + + + + + + Insulin-like growth factor 1 receptor (IGF1R), IGF-1R, type I IGF receptor (IGF-IR), CD1b R1, T6m Leu6 b-2-Microglobulin + + + – + – – – CD94 KLRD1, Kp43 HLA class I, NKG2-A, p39 + – + – – – – – – CD221 Insulin-like growth factor 1 (IGF-I), IGF-II, Insulin JTK13 + + + + + + + + + CD1c M241, R7, T6, Leu6, BDCA1 b-2-Microglobulin + + + – + – – – CD178, FASLG, APO-1, FAS, TNFRSF6, CD95L, APT1LG1, APT1, FAS1, FASTM, CD95 CD178 (Fas ligand) + + + + + – – IGF-II, TGF-b latency-associated peptide (LAP), Proliferin, Prorenin, Plasminogen, ALPS1A, TNFSF6, FASL Cation-independent mannose-6-phosphate receptor (M6P-R, CIM6PR, CIMPR, CI- CD1d R3G1, R3 b-2-Microglobulin, MHC II CD222 Leukemia -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

Integration Analysis for Novel Lncrna Markers Predicting Tumor Recurrence in Human Colon Adenocarcinoma Fangyao Chen1, Zhe Li2, Changyu Deng3 and Hong Yan1*
Chen et al. J Transl Med (2019) 17:299 https://doi.org/10.1186/s12967-019-2049-2 Journal of Translational Medicine RESEARCH Open Access Integration analysis for novel lncRNA markers predicting tumor recurrence in human colon adenocarcinoma Fangyao Chen1, Zhe Li2, Changyu Deng3 and Hong Yan1* Abstract Background: Numerous evidence has suggested that long non-coding RNA (lncRNA) acts an important role in tumor biology. This study focuses on the identifcation of novel prognostic lncRNA biomarkers predicting tumor recurrence in human colon adenocarcinoma. Methods: We obtained the research data from The Cancer Genome Atlas (TCGA) database. The interaction among diferent expressed lncRNA, miRNA and mRNA markers between colon adenocarcinoma patients with and without tumor recurrence were verifed with miRcode, starBase and miRTarBase databases. We established the lncRNA– miRNA–mRNA competing endogenous RNA (ceRNA) network based on the verifed association between the selected markers. We performed the functional enrichment analysis to obtain better understanding of the selected lncRNAs. Then we use multivariate logistic regression to identify the prognostic lncRNA markers with covariates. We also gener- ated a nomogram predicting tumor recurrence risk based on the identifed lncRNA biomarkers and clinical covariates. Results: We included 12,727 lncRNA, 1881 miRNA and 47,761 mRNA profling and clinical features for 113 colon adenocarcinoma patients obtained from the TCGA database. After fltration, we used 37 specifc lncRNAs, 60 miRNAs and 148 mRNAs in the ceRNA network analysis. We identifed fve lncRNAs as prognostic lncRNA markers predicting tumor recurrence in colon adenocarcinoma, in which four of them were identifed for the frst time. Finally, we gener- ated a nomogram illustrating the association between the identifed lncRNAs and the tumor recurrence risk in colon adenocarcinoma. -
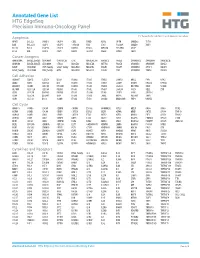
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1