Trigger Loop of RNA Polymerase Is a Positional, Not Acid–Base
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biomolecules
CHAPTER 3 Biomolecules 3.1 Carbohydrates In the previous chapter you have learnt about the cell and 3.2 Fatty Acids and its organelles. Each organelle has distinct structure and Lipids therefore performs different function. For example, cell membrane is made up of lipids and proteins. Cell wall is 3.3 Amino Acids made up of carbohydrates. Chromosomes are made up of 3.4 Protein Structure protein and nucleic acid, i.e., DNA and ribosomes are made 3.5 Nucleic Acids up of protein and nucleic acids, i.e., RNA. These ingredients of cellular organelles are also called macromolecules or biomolecules. There are four major types of biomolecules— carbohydrates, proteins, lipids and nucleic acids. Apart from being structural entities of the cell, these biomolecules play important functions in cellular processes. In this chapter you will study the structure and functions of these biomolecules. 3.1 CARBOHYDRATES Carbohydrates are one of the most abundant classes of biomolecules in nature and found widely distributed in all life forms. Chemically, they are aldehyde and ketone derivatives of the polyhydric alcohols. Major role of carbohydrates in living organisms is to function as a primary source of energy. These molecules also serve as energy stores, 2021-22 Chapter 3 Carbohydrade Final 30.018.2018.indd 50 11/14/2019 10:11:16 AM 51 BIOMOLECULES metabolic intermediates, and one of the major components of bacterial and plant cell wall. Also, these are part of DNA and RNA, which you will study later in this chapter. The cell walls of bacteria and plants are made up of polymers of carbohydrates. -

Site-Selective Artificial Ribonucleases: Renaissance of Oligonucleotide Conjugates for Irreversible Cleavage of RNA Sequences
molecules Review Site-Selective Artificial Ribonucleases: Renaissance of Oligonucleotide Conjugates for Irreversible Cleavage of RNA Sequences Yaroslav Staroseletz 1,†, Svetlana Gaponova 1,†, Olga Patutina 1, Elena Bichenkova 2 , Bahareh Amirloo 2, Thomas Heyman 2, Daria Chiglintseva 1 and Marina Zenkova 1,* 1 Laboratory of Nucleic Acids Biochemistry, Institute of Chemical Biology and Fundamental Medicine SB RAS, Lavrentiev’s Ave. 8, 630090 Novosibirsk, Russia; [email protected] (Y.S.); [email protected] (S.G.); [email protected] (O.P.); [email protected] (D.C.) 2 School of Health Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Oxford Rd., Manchester M13 9PT, UK; [email protected] (E.B.); [email protected] (B.A.); [email protected] (T.H.) * Correspondence: [email protected]; Tel.: +7-383-363-51-60 † These authors contributed equally to this work. Abstract: RNA-targeting therapeutics require highly efficient sequence-specific devices capable of RNA irreversible degradation in vivo. The most developed methods of sequence-specific RNA cleav- age, such as siRNA or antisense oligonucleotides (ASO), are currently based on recruitment of either intracellular multi-protein complexes or enzymes, leaving alternative approaches (e.g., ribozymes Citation: Staroseletz, Y.; Gaponova, and DNAzymes) far behind. Recently, site-selective artificial ribonucleases combining the oligonu- S.; Patutina, O.; Bichenkova, E.; cleotide recognition motifs (or their structural -

Molecular Biology and Applied Genetics
MOLECULAR BIOLOGY AND APPLIED GENETICS FOR Medical Laboratory Technology Students Upgraded Lecture Note Series Mohammed Awole Adem Jimma University MOLECULAR BIOLOGY AND APPLIED GENETICS For Medical Laboratory Technician Students Lecture Note Series Mohammed Awole Adem Upgraded - 2006 In collaboration with The Carter Center (EPHTI) and The Federal Democratic Republic of Ethiopia Ministry of Education and Ministry of Health Jimma University PREFACE The problem faced today in the learning and teaching of Applied Genetics and Molecular Biology for laboratory technologists in universities, colleges andhealth institutions primarily from the unavailability of textbooks that focus on the needs of Ethiopian students. This lecture note has been prepared with the primary aim of alleviating the problems encountered in the teaching of Medical Applied Genetics and Molecular Biology course and in minimizing discrepancies prevailing among the different teaching and training health institutions. It can also be used in teaching any introductory course on medical Applied Genetics and Molecular Biology and as a reference material. This lecture note is specifically designed for medical laboratory technologists, and includes only those areas of molecular cell biology and Applied Genetics relevant to degree-level understanding of modern laboratory technology. Since genetics is prerequisite course to molecular biology, the lecture note starts with Genetics i followed by Molecular Biology. It provides students with molecular background to enable them to understand and critically analyze recent advances in laboratory sciences. Finally, it contains a glossary, which summarizes important terminologies used in the text. Each chapter begins by specific learning objectives and at the end of each chapter review questions are also included. -

Nucleosides & Nucleotides
Nucleosides & Nucleotides Biochemistry Fundamentals > Genetic Information > Genetic Information NUCLEOSIDE AND NUCLEOTIDES SUMMARY NUCLEOSIDES  • Comprise a sugar and a base NUCLEOTIDES  • Phosphorylated nucleosides (at least one phosphorus group) • Link in chains to form polymers called nucleic acids (i.e. DNA and RNA) N-BETA-GLYCOSIDIC BOND  • Links nitrogenous base to sugar in nucleotides and nucleosides • Purines: C1 of sugar bonds with N9 of base • Pyrimidines: C1 of sugar bonds with N1 of base PHOSPHOESTER BOND • Links C3 or C5 hydroxyl group of sugar to phosphate NITROGENOUS BASES  • Adenine • Guanine • Cytosine • Thymine (DNA) 1 / 8 • Uracil (RNA) NUCLEOSIDES • =sugar + base • Adenosine • Guanosine • Cytidine • Thymidine • Uridine NUCLEOTIDE MONOPHOSPHATES – ADD SUFFIX 'SYLATE' • = nucleoside + 1 phosphate group • Adenylate • Guanylate • Cytidylate • Thymidylate • Uridylate Add prefix 'deoxy' when the ribose is a deoxyribose: lacks a hydroxyl group at C2. • Thymine only exists in DNA (deoxy prefix unnecessary for this reason) • Uracil only exists in RNA NUCLEIC ACIDS (DNA AND RNA)  • Phosphodiester bonds: a phosphate group attached to C5 of one sugar bonds with - OH group on C3 of next sugar • Nucleotide monomers of nucleic acids exist as triphosphates • Nucleotide polymers (i.e. nucleic acids) are monophosphates • 5' end is free phosphate group attached to C5 • 3' end is free -OH group attached to C3 2 / 8 FULL-LENGTH TEXT • Here we will learn about learn about nucleoside and nucleotide structure, and how they create the backbones of nucleic acids (DNA and RNA). • Start a table, so we can address key features of nucleosides and nucleotides. • Denote that nucleosides comprise a sugar and a base. -

DNA Wrap: Packaging Matters an Introduction to Epigenetics UNC-Chapel Hill’S Superfund Research Program
DNA Wrap: Packaging Matters An Introduction to Epigenetics UNC-Chapel Hill’s Superfund Research Program What causes the physical appearance and health status of identical twins to diverge with age? In this lesson, students learn that the environment can alter the way our genes are expressed, making even identical twins different. After watching a PBS video, A Tale of Two Mice, and reviewing data presented in the Environmental Health Perspectives article Maternal Genistein Alters Coat Color and Protects Avy Mouse Offspring from Obesity by Modifying the Fetal Epigenome, students learn about epigenetics and its role in regulating gene expression. Author Dana Haine, MS University of North Carolina at Chapel Hill Superfund Research Program Reviewers Dana Dolinoy, PhD University of Michigan Rebecca Fry, PhD University of North Carolina at Chapel Hill Superfund Research Program Banalata Sen, PhD, Audrey Pinto, PhD, Susan Booker, Dorothy Ritter Environmental Health Perspectives The funding for development of this lesson was provided by the National Institute of Environmental Health Sciences and the UNC Superfund Program. Learning Objectives By the end of this lesson students should be able to: define the term “epigenetics” describe DNA methylation as a mechanism for inhibiting gene transcription describe how gene expression can vary among genetically identical offspring Alignment to NC Essential Science Standards for Biology Bio.3.1 Explain how traits are determined by the structure and function of DNA. Bio.3.2.3 Explain how the environment can influence the expression of genetic traits. Bio.4.1 Understand how biological molecules are essential to the survival of living organisms. Bio.4.1.2 Summarize the relationship among DNA, proteins and amino acids in carrying out the work of cells and how this is similar in all organisms. -

Yeast Ribonuclease H(70) Cleaves RNA-DNA Junctions
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Volume 206, number 2 FEBS 4062 October 1986 Yeast ribonuclease H(70) cleaves RNA-DNA junctions Robert Karwan and Ulrike Wintersberger Department of Molecular GEnetics, Institute for Tumorbiology and Cancer Research, University of Vienna, Borschkegasse 8a, A-1090 Wien, Austria Received 7 August 1986 A specific substrate, Ml3 DNA:RNA-[3zP]DNA, was synthesized to investigate the mode of cleavage of enzymes with RNase H activity. RNase H(70) from Saccharomyces cerevisiae hydrolyzes the phosphodiester bond at the RNA-DNA junction of this substrate, thereby producing a 5’-monophosphate-terminated poly- deoxyribonucleotide and 3’-hydroxyl-terminated oligoribonucleotides. RNase H DNA-RNA hybrid DNA replication RNA primer removal (Saccharomyces cerevisiae) 1. INTRODUCTION ing short RNA primers. The phosphorus atom of the phosphate bridging the RNA and DNA parts Recently we have purified a ribonuclease H of this polynucleotide is radiolabeled (see fig.1). (RNase H, i.e. an enzyme which specifically Here we show that, in vitro the yeast RNase H(70) hydrolyzes the RNA strand of a DNA-RNA preferentially hydrolyzes the phosphodiester bond hybrid) from the yeast, Saccharomyces cerevisiae, between the ribonucleotide and the deox- which stimulates the in vitro DNA synthesis by yribonucleotide at the RNA-DNA junction. DNA polymerase A from the same organism [ 1,2]. Under certain conditions this protein also exhibits 2. MATERIALS AND METHODS reverse transcriptase activity [3]. As with RNases H from other organisms [4] no definite RNase H(70) and DNA polymerase A from S. -

212 Chapter 28 Biomolecules: Heterocycles and Nucleic Acids
Chapter 28 Biomolecules: Heterocycles and Nucleic Acids Heterocycles: cyclic organic compounds that contain rings atoms other than carbon (N,S,O are the most common). 28.1 Five-Membered Unsaturated Heterocycles (please read) 28.2 Structures of Pyrrole, Furan, and Thiophene (please read) 3 3 3 2 2 2 N 1 O 1 S 1 H Cyclopentadienyl Pyrole Furan Thiophene anion 28.3 Electrophilic Substitution Reactions of Pyrrole, Furan, and Thiophene (please read) 28.4 Pyridine, a Six-Membered Heterocycle (please read) 28.5 Electrophilic Substitution of Pyridine (please read) 28.6 Nucleophilic Substitution of Pyridine (please read) 28.7 Fused-Ring Heterocycles (please read) These sections contain some important concepts that were covered previously. 418 28.8 Nucleic Acids and Nucleotides Nucleic acids are the third class of biopolymers (polysaccharides and proteins being the others) Two major classes of nucleic acids deoxyribonucleic acid (DNA): carrier of genetic information ribonucleic acid (RNA): an intermediate in the expression of genetic information and other diverse roles The Central Dogma (F. Crick): DNA mRNA Protein (genome) (proteome) The monomeric units for nucleic acids are nucleotides Nucleotides are made up of three structural subunits 1. Sugar: ribose in RNA, 2-deoxyribose in DNA 2. Heterocyclic base 3. Phosphate 419 212 Nucleoside, nucleotides and nucleic acids phosphate sugar base phosphate phosphate sugar base sugar base sugar base phosphate nucleoside nucleotides sugar base nucleic acids The chemical linkage between monomer units in nucleic -

The Effect of Nucleic Acid Modifications on Digestion by DNA Exonucleases by Greg Lohman, Ph.D., New England Biolabs, Inc
be INSPIRED FEATURE ARTICLE drive DISCOVERY stay GENUINE The effect of nucleic acid modifications on digestion by DNA exonucleases by Greg Lohman, Ph.D., New England Biolabs, Inc. New England Biolabs offers a wide variety of exonucleases with a range of nucleotide structure specificity. Exonucleases can be active on ssDNA and/or dsDNA, initiate from the 5´ end and/or the 3´ end of polynucleotides, and can also act on RNA. Exonucleases have many applications in molecular biology, including removal of PCR primers, cleanup of plasmid DNA and production of ssDNA from dsDNA. In this article, we explore the activity of commercially available exonucleases on oligonucleotides that have chemical modifications added during phosphoramidite synthesis, including phosphorothioate diester bonds, 2´-modified riboses, modified bases, and 5´ and 3´ end modifications. We discuss how modifications can be used to selectively protect some polynucleotides from digestion in vitro, and which modifications will be cleaved like natural DNA. This information can be helpful for designing primers that are stable to exonucleases, protecting specific strands of DNA, and preparing oligonucleotides with modifications that will be resistant to rapid cleavage by common exonuclease activities. The ability of nucleases to hydrolyze phosphodi- of exonucleases available from NEB can be found Figure 1: ester bonds in nucleic acids is among the earliest in our selection chart, Common Applications of Examples of exonuclease directionality nucleic acid enzyme activities to be characterized Exonucleases and Non-specific Endonucleases, at → (1-6). Endonucleases cleave internal phosphodiester go.neb.com/ExosEndos. 3´ 5´ exonuclease bonds, while exonucleases, the focus of this article, What about cases where you only want to degrade 5´ 3´ must begin at the 5´ or 3´ end of a nucleic acid some of the ssDNA in a reaction? Or, when you 3´ 5´ strand and cleave the bonds sequentially (Figure 1). -

The Structure and Function of Large Biological Molecules by Dr. Ty C.M. Hoffman
The Structure and Function of Large Biological Molecules by Dr. Ty C.M. Hoffman Slide 1 All of the biological macromolecules are built from smaller subunits. Each subunit features -H and -OH substituents located somewhere on the subunit. This allows for assembly or disassembly of macromolecules via two classes of chemical reactions: • In a dehydration reaction, two subunits are joined after one subunit is stripped of -OH and the other subunit is stripped of -H. The -OH and the -H that are removed form H2O (water), which is why the reaction is so named. • In a hydrolysis reaction, a molecule is split into smaller particles. One of the smaller particles is supplied with -H to satisfy the valence; the other is supplied with -OH. The -H and -OH come from water, which is required for a hydrolysis reaction. Slide 2 Monosaccharides are simple (one subunit) sugars. Monosaccharides can be classified on the basis of the number of carbon atoms making up the skeleton or on the basis of where the carbonyl group is located in the skeleton (i.e., on whether the monosaccharide is an aldehyde or a ketone). Slide 3 Any monosaccharide can exist in either a linear form or a ring form, and the monosaccharide can be converted back and forth between the two forms. Making the ring requires breaking the double bond, so a monosaccharide features a carbonyl group only when it is in the linear form. Slide 4 When two monosaccharides are in the ring form, they can be bound together by a dehydration reaction. The covalent bond that ties together the monosaccharides is called a glycosidic linkage. -
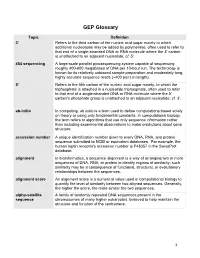
GEP Glossary
GEP Glossary Topic Definition 3' Refers to the third carbon of the nucleic acid sugar moiety to which additional nucleotides may be added by polymerase, often used to refer to that end of a single-stranded DNA or RNA molecule where the 3' carbon is unattached to an adjacent nucleotide; cf. 5'. 454 sequencing A large-scale parallel pyrosequencing system capable of sequencing roughly 400-600 megabases of DNA per 10-hour run. The technology is known for its relatively unbiased sample preparation and moderately long, highly accurate sequence reads (~400 pairs in length). 5' Refers to the fifth carbon of the nucleic acid sugar moiety, to which the triphosphate is attached in a nucleotide triphosphate, often used to refer to that end of a single-stranded DNA or RNA molecule where the 5' carbon's phosphate group is unattached to an adjacent nucleotide; cf. 3'. ab-initio In computing, ab initio is a term used to define computations based solely on theory or using only fundamental constants. In computational biology, the term refers to algorithms that use only sequence information rather than including experimental observations to make predictions about gene structure. accession number A unique identification number given to every DNA, RNA, and protein sequence submitted to NCBI or equivalent databases. For example, the human leptin receptor's accession number is P48357 in the SwissProt database. alignment In bioinformatics, a sequence alignment is a way of arranging two or more sequences of DNA, RNA, or protein to identify regions of similarity; such similarity may be a consequence of functional, structural, or evolutionary relationships between the sequences. -

DNA, RNA, Replication and Transcription
Harriet Wilson, Lecture Notes Bio. Sci. 4 - Microbiology Sierra College DNA, RNA, Replication and Transcription The metabolic processes described earlier (glycolysis, respiration, photophosphorylation, etc.), are dependent upon the enzymes present within cells. Most enzymes are proteins, (a few are RNA), and their presence within a cell is determined by the genetic information or hereditary material present. This material, contained primarily within the nucleus (eukaryotic cells) or nucleoid (prokaryotic cells), is deoxyribonucleic acid, commonly referred to as DNA. Background Information: According to a National Geographic article (Vol. 150 #3, 1976), the human body contains trillions of cells and each cell contains around 100,000 genes (segments of DNA). This amount of information, if written out, would fill around 600, 1000-page books (give or take a few as influenced by font size, paper weight, etc.). Within cells, the genetic information is tightly coiled, but if the DNA from all the cells within the human body were stretched out and laid end-to-end, it would extend to the sun and back over 400 times. This same amount of DNA would fit in a box about the size of an ice cube. DNA is amazing material with respect to its information storage potential. Composition of DNA: Deoxyribonucleic acid (DNA) is a polymer, i.e., a long, slender molecule composed of many, small, repeating units called nucleotides. Each cellular DNA molecule forms a double helix or duplex, i.e., includes two chains of nucleotides connected to one another by hydrogen bonds, and twisted into a helical configuration (like a twisted ladder). Each nucleotide (monomer) contains deoxyribose (a pentose monosaccharide or 5-carbon sugar), a phosphate group (PO4-) and one nitrogenous base. -

Synergism and Mutualism in Non-Enzymatic RNA Polymerization
Life 2014, 4, 598-620; doi:10.3390/life4040598 OPEN ACCESS life ISSN 2075-1729 www.mdpi.com/journal/life Review Synergism and Mutualism in Non-Enzymatic RNA Polymerization Hussein Kaddour and Nita Sahai * Department of Polymer Science, The University of Akron, Akron, OH 44325-3909, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-330-972-5795; Fax: +1-330-972-5290. External Editors: Fabio Mavelli and Pasquale Stano Received: 18 September 2014; in revised form: 15 October 2014 / Accepted: 17 October 2014 / Published: 3 November 2014 Abstract: The link between non-enzymatic RNA polymerization and RNA self-replication is a key step towards the “RNA world” and still far from being solved, despite extensive research. Clay minerals, lipids and, more recently, peptides were found to catalyze the non-enzymatic synthesis of RNA oligomers. Herein, a review of the main models for the formation of the first RNA polymers is presented in such a way as to emphasize the cooperation between life’s building blocks in their emergence and evolution. A logical outcome of the previous results is a combination of these models, in which RNA polymerization might have been catalyzed cooperatively by clays, lipids and peptides in one multi-component prebiotic soup. The resulting RNAs and oligopeptides might have mutualistically evolved towards functional RNAs and catalytic peptides, preceding the first RNA replication, thus supporting an RNA-peptide world. The investigation of such a system is a formidable challenge, given its complexity deriving from a tremendously large number of reactants and innumerable products.