HHS Public Access Author Manuscript
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Note ‐ Novel Genetic Loci for Atrial Fibrillation
Supplementary Note ‐ Novel genetic loci for atrial fibrillation SUPPLEMENTARY NOTE Large‐scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation INDEX 1. Supplementary tables Supplementary Table 1. Baseline characteristics GWAS Supplementary Table 2. Baseline characteristics ExWAS Supplementary Table 3. Detailed description of the genes at novel atrial fibrillation loci Supplementary Table 4. Results from Asian ancestry SKAT gene based test Supplementary Table 5. Single variant association results for the variants that were analyzed in the two significant gene‐based tests for SH3PDX2A in the Asian ancestry group Supplementary Table 6. Results from ancestry‐specific GWAS meta‐analyses Supplementary Table 7. Results from European and Asian ancestry ExWAS meta‐ analyses Supplementary Table 8. Results from European incident atrial fibrillation GWAS meta‐ analysis Supplementary Table 9. Results from European prevalent atrial fibrillation GWAS meta‐ analysis Supplementary Table 10. Comparison of results for common variant loci between the AFGen Consortium combined ancestry analysis and the Biobank Japan study Supplementary Table 11. Comparison of results for common variant loci between the AFGen Consortium combined ancestry analysis and the UK Biobank study. Supplementary Table 12. Approximate and joint conditional analysis in European ancestry results identify 20 independent genetic loci associated with atrial fibrillation Supplementary Table 13. Overlap with atrial fibrillation risk factor GWAS loci Supplementary Table 14. Association between novel atrial fibrillation loci and stroke subtypes in the Neuro‐CHARGE Stroke Consortium Supplementary Note ‐ Novel genetic loci for atrial fibrillation Supplementary Table 15. Association between novel atrial fibrillation loci and stroke subtypes in the Metastroke Consortium Supplementary Table 16. -
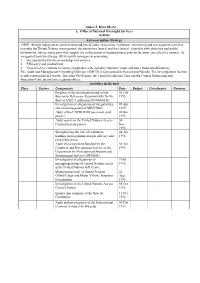
Annex 5. Data Sheets A. Office of Internal Oversight Services (OIOS)
Annex 5. Data Sheets a. Office of Internal Oversight Services (OIOS) Anti-corruption Strategy OIOS, through independent, professional and timely audit, inspection, evaluation, monitoring and investigation activities, provides the United Nations management, the Secretary-General and the General Assembly with objective and useful information, advice and reports that support the achievement of organizational goals in the most cost-effective manner. As an agent of positive change, OIOS assists managers in promoting: 1. Accountability for the stewardship of resources 2. Efficiency and productivity 3. Cost-effective controls to ensure compliance with authority minimise waste and deter fraud and dishonesty. The Audit and Management Consulting Division (AMCD) is represented in Geneva and Nairobi. The Investigations Section is only represented in Nairobi. The other OIOS units, the Central Evaluation Unit and the Central Monitoring and Inspection Unit, do not have regional offices. Activities in the past Place Sectors Components Date Budget Coordinator Partners Progress of the investigation and action 02 Feb thereon to Determine Responsibility for the 1995 theft of US$3.9 million in UNOSOM II Investigation of allegations of irregularities 05 Apr and mismanagement in MINURSO 1995 Audit of the UNPROFOR personnel pilot 06 Jun project 1995 Audit report on the United Nations Access 30 Control System project Nov 1995 Strengthening the role of evaluation 04 Apr findings in programme design, delivery and 1996 policy directives Audit of procurement handled -

For Peer Review Only
BMJ Open BMJ Open: first published as 10.1136/bmjopen-2014-005276 on 7 November 2014. Downloaded from FAST Warning Signs in Younger Stroke Patients: Implications for Stroke Education Campaigns ForJournal: peerBMJ Open review only Manuscript ID: bmjopen-2014-005276 Article Type: Research Date Submitted by the Author: 17-Mar-2014 Complete List of Authors: Kaps, M; JustusLiebig Universit�t Giessen Grittner, Ulrike; Department of Biostatistics and Clinical Epidemiology, Charité- University Medicine Berlin, Germany, Tanislav, Christian; Justus Liebig University , Neurology Jungehuelsing, Gerhard; Charit� Berlin, Department of Neurology Tatlisumak, Turgut; Helsinki University Central Hospital, Department of Neurology Kessler, Christof; University Greifswald, Neurology Schmidt, Reinhold; Department of Neurology, University of Graz, Austria, Putaala, Jukka; University of Helsinki, Department of Neurological Sciences; Helsinki University Central Hospital, Department of Neurology Norrving, Bo; Lund University Hospital Rolfs, Arndt; University of Rostock, Department of Neurology <b>Primary Subject http://bmjopen.bmj.com/ Health services research Heading</b>: Cardiovascular medicine, Diagnostics, Epidemiology, Health services Secondary Subject Heading: research, Neurology Stroke < NEUROLOGY, HEALTH SERVICES ADMINISTRATION & Keywords: MANAGEMENT, EPIDEMIOLOGY, MEDICAL EDUCATION & TRAINING, NEUROLOGY on September 25, 2021 by guest. Protected copyright. For peer review only - http://bmjopen.bmj.com/site/about/guidelines.xhtml Page 1 of 25 BMJ Open BMJ -

Genome-Wide Association Analysis of Young Onset Stroke Identifies a Locus on Chromosome 10Q25 Near HABP2
Original Contribution Genome-Wide Association Analysis of Young Onset Stroke Identifies a Locus on Chromosome 10q25 Near HABP2 Yu-Ching Cheng, PhD; Tara M. Stanne, PhD; Anne-Katrin Giese, MD; Weang Kee Ho, PhD; Matthew Traylor, PhD; Philippe Amouyel, MD, PhD; Elizabeth G. Holliday, PhD; Rainer Malik, PhD; Huichun Xu, MD, PhD; Steven J. Kittner, MD; John W. Cole, MD; Jeffrey R. O’Connell, PhD; John Danesh, DPhil, FRCP; Asif Rasheed, MBBS; Wei Zhao, MSc; Stefan Engelter, MD; Caspar Grond-Ginsbach, PhD; Yoichiro Kamatani, MD, PhD; Mark Lathrop, PhD; Didier Leys, MD, PhD; Vincent Thijs, MD, PhD; Tiina M. Metso, MD, PhD; Turgut Tatlisumak, MD, PhD; Alessandro Pezzini, MD; Eugenio A. Parati, MD; Bo Norrving, MD, PhD; Steve Bevan, PhD; Peter M. Rothwell, MD, PhD, FRCP; Cathie Sudlow, DPhil, FRCP; Agnieszka Slowik, MD, PhD; Arne Lindgren, MD, PhD; Matthew R. Walters, MD; WTCCC-2 Consortium; Jim Jannes, PhD; Jess Shen, MSc; David Crosslin, PhD; Kimberly Doheny, PhD; Cathy C. Laurie, PhD; Sandip M. Kanse, PhD; Joshua C. Bis, PhD; Myriam Fornage, PhD; Thomas H. Mosley, PhD; Jemma C. Hopewell, PhD; Konstantin Strauch, PhD; Martina Müller-Nurasyid, MD, PhD; Christian Gieger, PhD; Melanie Waldenberger, PhD, MPH; Annette Peters, PhD; Christine Meisinger, MD; M. Arfan Ikram, MD, PhD; W.T. Longstreth Jr, MD, MPH; James F. Meschia, MD; Sudha Seshadri, MD; Pankaj Sharma, PhD, MD; Bradford Worrall, MD, MSc; Christina Jern, MD, PhD; Christopher Levi, MBBS; Martin Dichgans, MD; Giorgio B. Boncoraglio, MD; Hugh S. Markus, MD; Stephanie Debette, MD, PhD; Arndt Rolfs, MD; Danish Saleheen, MBBS, PhD; Braxton D. Mitchell, PhD Received August 27, 2015; final revision received November 6, 2015; accepted November 18, 2015. -

Fabry Disease: a Review of Current Management Strategies
Revised submission June 2010 Fabry disease: a review of current management strategies A. MEHTA1, M. BECK2, F. EYSKENS3, C. FELICIANI4, I. KANTOLA5, U. RAMASWAMI6, A. ROLFS7, A. RIVERA8, S. WALDEK9, D.P. GERMAIN10 1Lysosomal Storage Disorders Unit, Department of Haematology, Royal Free Hospital and University College London School of Medicine, London, UK 2Children’s Hospital, University of Mainz, Mainz, Germany 3Department of Metabolic Diseases, ZNA Middelheim General Hospital, Antwerp, Belgium 4Department of Dermatology, Università Cattolica del Sacro Cuore, Rome, Italy 5Department of Medicine, Turku University Hospital, Turku, Finland 6Paediatric Metabolic Unit, Addenbrooke's Hospital, Cambridge, UK 7Albrecht-Kossel-Institute for Neuroregeneration, Medical Faculty, University of Rostock, Rostock, Germany 8Department of Internal Medicine, Complejo Hospitalario, Universitario de Vigo, Hospital Xeral de Vigo, Vigo, Spain 9Department of Adult Metabolic Diseases, Salford Royal NHS Foundation Trust, Salford, UK 10University of Versailles – St Quentin en Yvelines, Division of Medical Genetics, Hôpital Raymond Poincaré (AP-HP), 92380 Garches, France Corresponding author: Dr Atul Mehta, Lysosomal Storage Disorders Unit, Department of Academic Haematology, Royal Free and University College Medical School, Rowland Hill Street, London NW3 2PF, UK Tel: +44 (0) 20 7830 2814 Email: [email protected] Revised submission June 2010 Abstract Fabry disease is an X-linked inherited condition due to the absence or reduction of - galactosidase activity in lysosomes, that results in accumulation of globotriaosylceramide (Gb3) and related neutral glycosphingolipids. Manifestations of Fabry disease include serious and progressive impairment of renal and cardiac function. In addition, patients experience pain, gastrointestinal disturbance, transient ischaemic attacks and strokes. Additional effects on the skin, eyes, ears, lungs and bones are often seen. -
Ischaemic Stroke in Young Adults: a Global Perspective
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp-2019-322424 on 3 February 2020. Downloaded from Cerebrovascular disease REVIEW Ischaemic stroke in young adults: a global perspective Esther Boot,1 Merel Sanne Ekker ,1 Jukka Putaala,2 Steven Kittner,3 Frank-Erik De Leeuw,1 Anil M Tuladhar 1 ► Additional material is ABSTRact Furthermore, we will review the more traditional published online only. To view Ischaemic stroke at young age is an increasing problem vascular risk factors and geographical and racial/ please visit the journal online ethnic differences in their prevalence and strength (http:// dx. doi. org/ 10. 1136/ in both developing and developed countries due to jnnp- 2019- 322424). rising incidence, high morbidity and mortality and long- of association with stroke. term psychological, physical and social consequences. 1Department of Neurology, Compared with stroke in older adults, stroke in young METHODS Radboud Univerisity Medical adults is more heterogeneous due to the wide variety Centre, Donders Institute for For this review, we searched in MEDLINE and Brain, Cognition and Behaviour, of possible underlying risk factors and aetiologies. In WHO Database for articles published from 1 Centre for Neuroscience, this review, we will provide an overview of the global January 2008 to 1 January 2020. The following Nijmegen, The Netherlands variation in the epidemiology of stroke in young adults, 2 search terms (including MedicalSubject Headings) Department of Neurology, with special attention to differences in geography, Helsinki University Central were used in multiple combinations: ‘ischemic Hospital, Helsinki, Finland ethnicity/race and sex, as well as traditional and novel stroke, young adults, incidence, global, epidemi- 3 Department of Neurology, risk factors for early- onset ischaemic stroke, such ology, infection, sex differences, race, ethnicity, University of Maryland School of as air pollution. -

Clinically Relevant Depressive Symptoms in Young Stroke Patients – Results of the Sifap1 Study
Original Paper Neuroepidemiology 2015;44:30–38 Received: September 28, 2014 DOI: 10.1159/000371389 Accepted: December 4, 2014 Published online: January 31, 2015 Clinically Relevant Depressive Symptoms in Young Stroke Patients – Results of the sifap1 Study a b c, d c Christian Tanislav Peter Kropp Ulrike Grittner Martin Holzhausen e d f Franz Fazekas Gerhard Jan Jungehülsing Turgut Tatlisumak g f h i Bettina von Sarnowski Jukka Putaala Roman Huber Vincent Thijs e a e, j k Reinhold Schmidt Manfred Kaps Christian Enzinger Martin Dichgans l m Bo Norrving Arndt Rolfs a b Neurology, Justus Liebig University, Giessen , Institute of Medical Psychology and Medical Sociology, University c of Rostock, Rostock , Department for Biostatistics and Clinical Epidemiology, Charité-Universitätsmedizin Berlin, d e and Center for Stroke Research, Charité-Universitätsmedizin Berlin, Berlin , Germany; Department of Neurology, f Medical University of Graz, Graz , Austria; Department of Neurology, Helsinki University Central Hospital, g h Helsinki , Finland; Department of Neurology, University of Greifswald, Greifswald , Department of Neurology, i j Friedrichshafen , Germany; Clinical and Experimental Neurology, Leuven , Belgium; Department of Radiology, k Division of Neuroradiology, Medical University of Graz, Graz , Austria; Institute for Stroke and Dementia Research, l Klinikum Grosshadern, Munich , Germany; Department of Clinical Neuroscience, Neurology, Lund University, Lund , m Sweden; Albrecht-Kossel-Institute for Neuroregeneration, University of Rostock, Rostock , Germany Key Words relevant depressive symptoms (CRDS, defined by a BDI- Young stroke · Depression · Acute cerebral ischemia · score ≥ 18) the self-reporting Beck Depression Inventory sifap1 · Risk factors (BDI) was obtained on inclusion in the study. Associations with baseline parameters, stroke severity (National Institutes of Health Stroke Scale, NIHSS), and brain MRI findings were Abstract analyzed. -

FOR OFFICIAL USE ONLY Public Disclosure Authorized Report No
Document of The World Bank FOR OFFICIAL USE ONLY Public Disclosure Authorized Report No. 52691-MZ PROJECT APPRAISAL DOCUMENT ON A PROPOSED CREDIT Public Disclosure Authorized IN THE AMOUNT OF SDR 19.6 MILLION (US$30.4 MILLION EQUIVALENT) TO THE REPUBLIC OF MOZAMBIQUE FOR A NATIONAL DECENTRALIZED PLANNING AND FINANCE PROJECT Public Disclosure Authorized March 4,2010 Urban and Water Country Department AFCS2 Africa Region Public Disclosure Authorized This document has a restricted distribution and may be used by recipients only in the performance of their official duties. Its contents may not otherwise be disclosed without World Bank authorization. CURRENCY EQUIVALENTS (Exchange Rate Effective as January 3 1,201 0) Currency Unit = New Meticais (Mtn) 31.07Mtn = US$l US$1.55 = SDR1 FISCAL YEAR January 1 - December 31 ABBREVIATIONS AND ACRONYMS AD Administraqclo Distrital (District Administration) AT Assistzncia Te'cnica(Technical Assistance) AWPB Annual Work Plan and Budget BdPES Balanqo do Plano Econdmico Social (Monitoring System of the Annual Government Plan) CCD Conselho Consultivo Distrital (District Consultative Council) CdS Comite' de Supewisclo (Supervision Committee) CdSP Comite' de Supervisclo Provincial (Provincial Supervision Committee) CFAA Country Financial Accountability Assessment CFFM Common Fund Financing Mechanism CL Conselho Locais-CLs (Local Councils) CNCS Conselho Nacinal de Combate ao HIV/SIDA (National AIDS Council) CPAR Country Procurement Assessment Report CPS Country Partnership Strategy CQS Consultants' Qualification -

Catalysing Ocean Finance
Empowered lives. Resilient nations. Catalysing Ocean Finance Volume II Methodologies and Case Studies United Nations Development Programme Empowered lives. Resilient nations. UNDP partners with people at all levels of society to help build nations that can withstand crisis, and drive and sustain the kind of growth that improves the quality of life for everyone. On the ground in 177 countries and territories, we offer global perspective and local insight to help empower lives and build resilient nations. www.undp.org The GEF unites 182 countries in partnership with international institutions, non-governmental organizations (NGOs), and the private sector to address global environmental issues while supporting national sustainable development initiatives. Today the GEF is the largest public funder of projects to improve the global environment. An independently operating financial organization, the GEF provides grants for projects related to biodiversity, climate change, international waters, land degradation, the ozone layer, and persistent organic pollutants. Since 1991, GEF has achieved a strong track record with developing countries and countries with economies in transition, providing $9.2 billion in grants and leveraging $40 billion in co-financing for over 2,700 projects in over 168 countries. www.thegef.org September 2012 Catalysing Ocean Finance Volume II Methodologies and Case Studies Copyright © 2012 United Nations Development Programme United Nations Development Programme Global Environment Facility Bureau of Development Policy GEF Secretariat 304 East 45th Street , 1818 H Street, NW 9th Floor MSN P4-400 New York, NY 10017, USA Washington CD 20433, USA www.undp.org www.thegef.org All right reserved. This publication or parts of it may not be reproduced, stored by means of any system or transmitted, in any form or by any medium, whether electronic, mechanical, photocopied, recorded or of any other type, without the prior permission of the United Nations Development Programme. -

Acute Cerebrovascular Disease in the Young the Stroke in Young Fabry Patients Study
Acute Cerebrovascular Disease in the Young The Stroke in Young Fabry Patients Study Rolfs, Arndt; Fazekas, Franz; Grittner, Ulrike; Dichgans, Martin; Martus, Peter; Holzhausen, Martin; Boettcher, Tobias; Heuschmann, Peter U.; Tatlisumak, Turgut; Tanislav, Christian; Jungehulsing, Gerhard J.; Giese, Anne-Katrin; Putaala, Jukaa; Huber, Roman; Bodechtel, Ulf; Lichy, Christoph; Enzinger, Christian; Schmidt, Reinhold; Hennerici, Michael G.; Kaps, Manfred; Kessler, Christof; Lackner, Karl; Paschke, Eduard; Meyer, Wolfgang; Mascher, Hermann; Riess, Olaf; Kolodny, Edwin; Norrving, Bo Published in: Stroke: a journal of cerebral circulation DOI: 10.1161/STROKEAHA.112.663708 2013 Link to publication Citation for published version (APA): Rolfs, A., Fazekas, F., Grittner, U., Dichgans, M., Martus, P., Holzhausen, M., Boettcher, T., Heuschmann, P. U., Tatlisumak, T., Tanislav, C., Jungehulsing, G. J., Giese, A-K., Putaala, J., Huber, R., Bodechtel, U., Lichy, C., Enzinger, C., Schmidt, R., Hennerici, M. G., ... Norrving, B. (2013). Acute Cerebrovascular Disease in the Young The Stroke in Young Fabry Patients Study. Stroke: a journal of cerebral circulation, 44(2), 340-349. https://doi.org/10.1161/STROKEAHA.112.663708 Total number of authors: 28 General rights Unless other specific re-use rights are stated the following general rights apply: Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise -

THE CONTRIBUTIONS of MARINE and COASTAL AREA-BASED MANAGEMENT APPROACHES to SUSTAINABLE DEVELOPMENT GOALS and TARGETS Supplementary Annex
THE CONTRIBUTIONS OF MARINE AND COASTAL AREA-BASED MANAGEMENT APPROACHES TO SUSTAINABLE DEVELOPMENT GOALS AND TARGETS Supplementary Annex UN ENVIRONMENT REGIONAL SEAS REPORTS AND STUDIES NO. 205 Contents Introduction .................................................................................................................................................... 3 Case Study 1. Area of Particular Environmental Interest: Clarion-Clipperton Zone Areas of Particular Environmental Interest, Mid Pacific .............................................................................................................. 6 Case Study 2. Fisheries Closure and Conservation Area: Lyme Bay Reserve, United Kingdom ............... 16 Case Study 3. Integrated Coastal Zone Management: Coastal and Marine Spatial Planning in Belize ... 24 Case Study 4. Integrated Coastal Zone Management: Patagonian Coastal Zone Management Plan, Argentina ...................................................................................................................................................... 33 Case Study 5. Integrated Coastal Zone Management: Mediterranean Integrated Coastal Zone Management Protocol ................................................................................................................................. 43 Case Study 6. Large Marine Ecosystem: Bay of Bengal Large Marine Ecosystem Project (BOBLME)..... 54 Case Study 7. Locally Managed Marine Area: Madagascar Locally Managed Marine Area Network (MIHARI) ...................................................................................................................................................... -

Clinically Relevant Depressive Symptoms in Young Stroke Patients – Results of the Sifap1 Study
Original Paper Neuroepidemiology 2015;44:30–38 Received: September 28, 2014 DOI: 10.1159/000371389 Accepted: December 4, 2014 Published online: January 31, 2015 Clinically Relevant Depressive Symptoms in Young Stroke Patients – Results of the sifap1 Study a b c, d c Christian Tanislav Peter Kropp Ulrike Grittner Martin Holzhausen e d f Franz Fazekas Gerhard Jan Jungehülsing Turgut Tatlisumak g f h i Bettina von Sarnowski Jukka Putaala Roman Huber Vincent Thijs e a e, j k Reinhold Schmidt Manfred Kaps Christian Enzinger Martin Dichgans l m Bo Norrving Arndt Rolfs a b Neurology, Justus Liebig University, Giessen , Institute of Medical Psychology and Medical Sociology, University c of Rostock, Rostock , Department for Biostatistics and Clinical Epidemiology, Charité-Universitätsmedizin Berlin, d e and Center for Stroke Research, Charité-Universitätsmedizin Berlin, Berlin , Germany; Department of Neurology, f Medical University of Graz, Graz , Austria; Department of Neurology, Helsinki University Central Hospital, g h Helsinki , Finland; Department of Neurology, University of Greifswald, Greifswald , Department of Neurology, i j Friedrichshafen , Germany; Clinical and Experimental Neurology, Leuven , Belgium; Department of Radiology, k Division of Neuroradiology, Medical University of Graz, Graz , Austria; Institute for Stroke and Dementia Research, l Klinikum Grosshadern, Munich , Germany; Department of Clinical Neuroscience, Neurology, Lund University, Lund , m Sweden; Albrecht-Kossel-Institute for Neuroregeneration, University of Rostock, Rostock , Germany Key Words relevant depressive symptoms (CRDS, defined by a BDI- Young stroke · Depression · Acute cerebral ischemia · score ≥ 18) the self-reporting Beck Depression Inventory sifap1 · Risk factors (BDI) was obtained on inclusion in the study. Associations with baseline parameters, stroke severity (National Institutes of Health Stroke Scale, NIHSS), and brain MRI findings were Abstract analyzed.