Multiple Benzene-Formation Paths in a Fuel-Rich Cyclohexane Flame
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
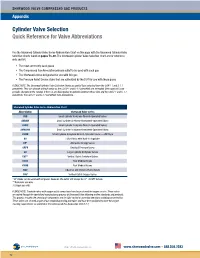
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

A Comparative Study of the Formation of Aromatics In
A COMPARATIVE STUDY OF THE FORMATION OF AROMATICS IN RICH METHANE FLAMES DOPED BY UNSATURATED COMPOUNDS H.A. GUENICHE, J. BIET, P.A. GLAUDE, R. FOURNET, F. BATTIN-LECLERC* Département de Chimie-Physique des Réactions, Nancy Université, CNRS, 1 rue Grandville, BP 20451, 54001 NANCY Cedex, France * E-mail : [email protected] ; Tel.: 33 3 83 17 51 25 , Fax : 33 3 83 37 81 20 ABSTRACT For a better modeling of the importance of the different channels leading to the first aromatic ring, we have compared the structures of laminar rich premixed methane flames doped with several unsaturated hydrocarbons: allene and propyne, because they are precursors of propargyl radicals which are well known as having an important role in forming benzene, 1,3-butadiene to put in evidence a possible production of benzene due to reactions of C4 compounds, and, finally, cyclopentene which is a source of cyclopentadienylmethylene radicals which in turn are expected to easily isomerizes to give benzene. These flames have been stabilized on a burner at a pressure of 6.7 kPa (50 Torr) using argon as dilutant, for equivalence ratios (φ) from 1.55 to 1.79. A unique mechanism, including the formation and decomposition of benzene and toluene, has been used to model the oxidation of allene, propyne, 1,3-butadiene and cyclopentene. The main reaction pathways of aromatics formation have been derived from reaction rate and sensitivity analyses and have been compared for the three types of additives. These combined analyses and comparisons can only been performed when a unique mechanism is available for all the studied additives. -

High Purity/Recovery Separation of Propylene from Propyne Using Anion Pillared Metal-Organic Framework: Application of Vacuum Swing Adsorption (VSA)
energies Article High Purity/Recovery Separation of Propylene from Propyne Using Anion Pillared Metal-Organic Framework: Application of Vacuum Swing Adsorption (VSA) Majeda Khraisheh 1, Fares AlMomani 1,* and Gavin Walker 2 1 Department of Chemical Engineering, College of Engineering, Qatar University, Doha P.O. Box 2713, Qatar; [email protected] 2 Department of Chemical Sciences, SSPC, Bernal Institute, University of Limerick, V94 T9PX Limerick, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +974-4403-4140 Abstract: Propylene is one of the world’s most important basic olefin raw material used in the produc- tion of a vast array of polymers and other chemicals. The need for high purity grade of propylene is essential and traditionally achieved by the very energy-intensive cryogenic separation. In this study, 2− a pillared inorganic anion SIF6 was used as a highly selective C3H4 due to the square grid pyrazine- based structure. Single gas adsorption revealed a very high C3H4 uptake value (3.32, 3.12, 2.97 and 2.43 mmol·g−1 at 300, 320, 340 and 360 K, respectively). The values for propylene for the same tempera- tures were 2.73, 2.64, 2.31 and 1.84 mmol·g−1, respectively. Experimental results were obtained for the two gases fitted using Langmuir and Toth models. The former had a varied degree of representation of the system with a better presentation of the adsorption of the propylene compared to the propyne system. The Toth model regression offered a better fit of the experimental data over the entire range of pressures. -

(12) United States Patent (10) Patent No.: US 8,198.491 B2 Masatoshi Et Al
USOO8198491 B2 (12) United States Patent (10) Patent No.: US 8,198.491 B2 Masatoshi et al. (45) Date of Patent: Jun. 12, 2012 (54) PROCESS FOR PREPARING 5,986,151 A 1 1/1999 Van Der Puy 2,3,3,3-TETRAFLUOROPROPENE AND $425, 23.99 Esthet al. 1,3,3,3-TETRAFLUOROPROPENE 7,833.434 B2 11/2010 Rao et al. 2005/0245773 Al 1 1/2005 Mukhopadhyay et al. (75) Inventors: Nose Masatoshi, Settsu (JP): Komatsu 2006/0258891 A1 1 1/2006 Mukhopadhyay et al. Yuzo, Settsu (JP); Sugiyama Akinari, 3.s! .k A. 239. SgO et stal al. Situ (JP); Shibanuma Takashi, Settsu 2009,0264689 A1 10, 2009 Rao et al. (JP) 2010/0200798 A1 8, 2010 Rao et al. (73) Assignee: Daikin Industries, Ltd., Osaka (JP) FOREIGN PATENT DOCUMENTS EP O 974 571 1, 2000 (*) Notice: Subject to any disclaimer, the term of this JP 63-211245 9, 1988 patent is extended or adjusted under 35 JP 11-14.0002 5, 1999 U.S.C. 154(b) by 0 days. JP 2007-320896 12/2007 WO 2008/OO2499 1, 2008 WO 2008/OO2500 1, 2008 (21) Appl. No.: 13/057,525 WO 2008/030440 3, 2008 (22) PCT Filed: Jul. 21, 2009 OTHER PUBLICATIONS International Search Report issued Mar. 3, 2010 in International (86). PCT No.: PCT/UP2O09/063314 (PCT) Application No. PCT/JP2009/063314. S371 (c)(1) PCT Written Opinion of the International Searching Authority issued (2), (4) Date:s Feb. 4, 2011 Mar.sia. 3, 2010 in International ((PCT) ) Application NoNo. PCT/JP2009/ Haszeldine et al., “Addition of FreeRadicals to Unsaturated Systems. -

Identification of a Simplest Hypervalent Hydrogen Fluoride
www.nature.com/scientificreports OPEN Identification of a Simplest Hypervalent Hydrogen Fluoride Anion in Solid Argon Received: 19 December 2016 Meng-Chen Liu1, Hui-Fen Chen2, Chih-Hao Chin1, Tzu-Ping Huang1, Yu-Jung Chen3 & Yu-Jong Accepted: 18 April 2017 Wu1,4 Published: xx xx xxxx Hypervalent molecules are one of the exceptions to the octet rule. Bonding in most hypervalent molecules is well rationalized by the Rundle–Pimentel model (three-center four-electron bond), and high ionic bonding between the ligands and the central atom is essential for stabilizing hypervalent molecules. Here, we produced one of the simplest hypervalent anions, HF−, which is known to deviate from the Rundle–Pimentel model, and identified its ro-vibrational features. High-levelab inito calculations reveal that its bond dissociation energy is comparable to that of dihalides, as supported by secondary photolysis experiments with irradiation at various wavelengths. The charge distribution analysis suggested that the F atom of HF− is negative and hypervalent and the bonding is more covalent than ionic. The octet rule indicates that atoms of the main group elements tend to gain or lose electrons in order to have eight electrons in their valence shells1, 2, similar to the electronic configuration of the noble gases. Therefore, to attain this fully filled electronic configuration, atoms combine to form molecules by sharing their valence electrons to form chemical bonds. This rule is especially applicable to the period 2 and 3 elements. Most molecules are formed by following this rule and the bonding structure of molecules can be easily recognized by using Lewis electron dot diagrams. -

Hydrogen Sulfide Capture: from Absorption in Polar Liquids to Oxide
Review pubs.acs.org/CR Hydrogen Sulfide Capture: From Absorption in Polar Liquids to Oxide, Zeolite, and Metal−Organic Framework Adsorbents and Membranes Mansi S. Shah,† Michael Tsapatsis,† and J. Ilja Siepmann*,†,‡ † Department of Chemical Engineering and Materials Science, University of Minnesota, 421 Washington Avenue SE, Minneapolis, Minnesota 55455-0132, United States ‡ Department of Chemistry and Chemical Theory Center, University of Minnesota, 207 Pleasant Street SE, Minneapolis, Minnesota 55455-0431, United States ABSTRACT: Hydrogen sulfide removal is a long-standing economic and environ- mental challenge faced by the oil and gas industries. H2S separation processes using reactive and non-reactive absorption and adsorption, membranes, and cryogenic distillation are reviewed. A detailed discussion is presented on new developments in adsorbents, such as ionic liquids, metal oxides, metals, metal−organic frameworks, zeolites, carbon-based materials, and composite materials; and membrane technologies for H2S removal. This Review attempts to exhaustively compile the existing literature on sour gas sweetening and to identify promising areas for future developments in the field. CONTENTS 4.1. Polymeric Membranes 9785 4.2. Membranes for Gas−Liquid Contact 9786 1. Introduction 9755 4.3. Ceramic Membranes 9789 2. Absorption 9758 4.4. Carbon-Based Membranes 9789 2.1. Alkanolamines 9758 4.5. Composite Membranes 9790 2.2. Methanol 9758 N 5. Cryogenic Distillation 9790 2.3. -Methyl-2-pyrrolidone 9758 6. Outlook and Perspectives 9791 2.4. Poly(ethylene glycol) Dimethyl Ether 9759 Author Information 9792 2.5. Sulfolane and Diisopropanolamine 9759 Corresponding Author 9792 2.6. Ionic Liquids 9759 ORCID 9792 3. Adsorption 9762 Notes 9792 3.1. -

Rich Methane Premixed Laminar Flames Doped with Light Unsaturated
Combustion and Flame 146 (2006) 620–634 www.elsevier.com/locate/combustflame Rich methane premixed laminar flames doped with light unsaturated hydrocarbons I. Allene and propyne H.A. Gueniche, P.A. Glaude, G. Dayma, R. Fournet, F. Battin-Leclerc ∗ Département de Chimie-Physique des Réactions, Nancy University, CNRS, ENSIC, 1 rue Grandville, BP 20451, 54001 Nancy Cedex, France Received 9 February 2006; received in revised form 16 June 2006; accepted 3 July 2006 Available online 4 August 2006 Abstract The structure of three laminar premixed rich flames has been investigated: a pure methane flame and two methane flames doped by allene and propyne, respectively. The gases of the three flames contain 20.9% (mo- lar) of methane and 33.4% of oxygen, corresponding to an equivalence ratio of 1.25 for the pure methane flame. In both doped flames, 2.49% of C3H4 was added, corresponding to a ratio C3H4/CH4 of 12% and an equivalence ra- tio of 1.55. The three flames have been stabilized on a burner at a pressure of 6.7 kPa using argon as dilutant, with a gas velocity at the burner of 36 cm/s at 333 K. The concentration profiles of stable species were measured by gas chromatography after sampling with a quartz microprobe. Quantified species included carbon monoxide and diox- ide, methane, oxygen, hydrogen, ethane, ethylene, acetylene, propyne, allene, propene, propane, 1,2-butadiene, 1,3-butadiene, 1-butene, isobutene, 1-butyne, vinylacetylene, and benzene. The temperature was measured using a PtRh (6%)–PtRh (30%) thermocouple settled inside the enclosure and ranged from 700 K close to the burner up to 1850 K. -

Common Name: METHYL ACETYLENE HAZARD SUMMARY
Common Name: METHYL ACETYLENE CAS Number: 74-99-7 RTK Substance number: 1218 DOT Number: UN 1954 Date: May 1999 ----------------------------------------------------------------------- ----------------------------------------------------------------------- HAZARD SUMMARY * Methyl Acetylene can affect you when breathed in. * Exposure to hazardous substances should be routinely * Breathing Methyl Acetylene can irritate the nose and evaluated. This may include collecting personal and area throat. air samples. You can obtain copies of sampling results * Breathing Methyl Acetylene can irritate the lungs causing from your employer. You have a legal right to this coughing and/or shortness of breath. Higher exposures information under OSHA 1910.1020. can cause a build-up of fluid in the lungs (pulmonary * If you think you are experiencing any work-related health edema), a medical emergency, with severe shortness of problems, see a doctor trained to recognize occupational breath. diseases. Take this Fact Sheet with you. * Exposure to Methyl Acetylene can cause headache, dizziness, convulsions (fits) and unconsciousness. WORKPLACE EXPOSURE LIMITS * Liquified Methyl Acetylene can cause frostbite. OSHA: The legal airborne permissible exposure limit * Methyl Acetylene is a HIGHLY FLAMMABLE and (PEL) is 1,000 ppm averaged over an 8-hour REACTIVE chemical and is a DANGEROUS FIRE and workshift. EXPLOSION HAZARD. NIOSH: The recommended airborne exposure limit is IDENTIFICATION 1,000 ppm averaged over a 10-hour workshift. Methyl Acetylene is a colorless gas, which is shipped as a liquid, with a sweet odor. It is used as a simple anesthetic and ACGIH: The recommended airborne exposure limit is in the manufacture of other chemicals, and as a welding torch 1,000 ppm averaged over an 8-hour workshift. -

H Atom Attack on Propene Claudette M
ARTICLE pubs.acs.org/JPCA H Atom Attack on Propene Claudette M. Rosado-Reyes,* Jeffrey A. Manion, and Wing Tsang National Institute of Standards and Technology, 100 Bureau Drive, Gaithersburg, Maryland 20899, United States bS Supporting Information d ABSTRACT: The reaction of propene (CH3CH CH2) with hydrogen atoms has been investigated in a heated single-pulsed shock tube at temperatures between 902 and 1200 K and pressures of 1.5À3.4 bar. Stable products from H atom addition and H abstraction have been identified and quantified by gas chromatography/flame ionization/mass spectrometry. The reaction for the H addition channel involving methyl displacement from propene has been determined relative to methyl displacement from 1,3,5-trimethylbenzene þ f d þ (135TMB), leading to a reaction rate, k(H propene) H2C CH2 Â 13 À 3 CH3) = 4.8 10 exp( 2081/T)cm/(mol s). The rate constant for the abstraction of the allylic hydrogen atom is determined to be k(H þ propene f d þ Â 13 À 3 CH2CH CH2 H2) = 6.4 10 exp( 4168/T)cm /(mol s). The reaction of H þ propene has also been directly studied relative to the reaction of H þ propyne, and the relationship is found to be log[k(H þ propyne f acetylene þ þ f þ À ( þ CH3)/k(H propene ethylene CH3)]=( 0.461 0.041)(1000/T) (0.44 ( 0.04). The results showed that the rate constant for the methyl displacement reaction with propene is a factor of 1.05 ( 0.1 larger than that for propyne near 1000 K. -

Propyne Safety Data Sheet 1500101 According to Federal Register / Vol
Propyne Safety Data Sheet 1500101 according to Federal Register / Vol. 77, No. 58 / Monday, March 26, 2012 / Rules and Regulations Date of issue: 11/01/2018 Version: 1.0 SECTION 1: Identification 1.1. Identification Product form : Substance Substance name : Propyne CAS No : 74-99-7 Product code : 1500-1-01 Formula : C3H4 Synonyms : Methylacetylene Other means of identification : MFCD00036235 1.2. Relevant identified uses of the substance or mixture and uses advised against Use of the substance/mixture : Laboratory chemicals Manufacture of substances Scientific research and development 1.3. Details of the supplier of the safety data sheet SynQuest Laboratories, Inc. P.O. Box 309 Alachua, FL 32615 - United States of America T (386) 462-0788 - F (386) 462-7097 [email protected] - www.synquestlabs.com 1.4. Emergency telephone number Emergency number : (844) 523-4086 (3E Company - Account 10069) SECTION 2: Hazard(s) identification 2.1. Classification of the substance or mixture Classification (GHS-US) Simple Asphy H380 - May displace oxygen and cause rapid suffocation Flam. Gas 1 H220 - Extremely flammable gas Liquefied gas H280 - Contains gas under pressure; may explode if heated STOT SE 3 H335 - May cause respiratory irritation Full text of H-phrases: see section 16 2.2. Label elements GHS-US labeling Hazard pictograms (GHS-US) : GHS02 GHS04 GHS07 Signal word (GHS-US) : Danger Hazard statements (GHS-US) : H220 - Extremely flammable gas H280 - Contains gas under pressure; may explode if heated H335 - May cause respiratory irritation H380 - May displace oxygen and cause rapid suffocation Precautionary statements (GHS-US) : P210 - Keep away from heat/sparks/open flames/hot surfaces. -

Catalytic Preparation of 2-Halo-1-Alkenes from Unsaturated
Europaisches Patentamt J European Patent Office © Publication number: 0 176 616 Office europeen des brevets A1 © EUROPEAN PATENT APPLICATION @ Application number: 84111837.5 © Int. CI.*: C 07 C 21/02 C 07 C 17/08 © Date of filing: 03.10.84 © Date of publication of application: © Applicant: THE DOW CHEMICAL COMPANY 09.04.86 Bulletin 86/15 2030 Dow Center Abbott Road P.O. Box 1967 Midland, MI48640IUS) © Designated Contracting States: BE DE FR GB IT NL @ Inventor: Klun, Robert T. 4161 E.Baker Road Route 11 Midland Michigan 48640IUS) © Inventor: Murchison, Craig B. 606 West Meadowbrook Midland Michigan 48640{US) © Inventor: Hucul, Dennis A. 5001 Foxcroft Midland Michigan 48640(US) © Representative: Weickmann, Heinrich, Dipl.-lng. et al, Patentanwalte Dipl.-lng. H.Weickmann Dipl.-Phys.Dr. K.Rncke Dipl.-lng. F.A.Weickmann Dipl.-Chem. B. Huber Dr.-ing. H. Liska Dipl.-Phys.Dr. J. Prechtel Postfach 860820 D-8000 Munchen 86(DE) © Catalytic preparation of 2-halo-1-alkenesfrom unsaturated hydrocarbons. 57© AA 2-halo-1-alkene (such as 2-chloro-1-propene) is pre- pared by contacting together, in the presence of a catalyst (such as alumina), (a) a 1,2-diene (such as propadiene) and/or a 1-alkyne (such as propyne), with (b) a hydrogen halide (such as HCI); in the presence of at least 1,000 ppm water. The process of the invention provides unexpected selectivity for the desired product. Croydon Printing Company Ltd The invention relates to an improved process for the preparation of 2-halo-l-alkenes from light hydro- carbon streams. Griesbaum, US 3,377,389, April 9, 1968, teaches that hydrogen chloride and hydrogen bromide will add to unsaturated compounds in accordance with Markovnikov's Rule and that allene (propadiene) and methylacetylene (propyne) are hydrohalogenated to give 2-halopropene. -

Polycyclic Aromatic Hydrocarbons in Pyrolysis of Gasoline Surrogates ( N -Heptane/ Iso -Octane/Toluene)
Polycyclic aromatic hydrocarbons in pyrolysis of gasoline surrogates ( n -heptane/ iso -octane/toluene) Item Type Article Authors Shao, Can; Wang, Haoyi; Atef, Nour; Wang, Zhandong; Chen, Bingjie; Almalki, Maram M.; Zhang, Yan; Cao, Chuangchuang; Yang, Jiuzhong; Sarathy, Mani Citation Shao C, Wang H, Atef N, Wang Z, Chen B, et al. (2018) Polycyclic aromatic hydrocarbons in pyrolysis of gasoline surrogates ( n - heptane/ iso -octane/toluene). Proceedings of the Combustion Institute. Available: http://dx.doi.org/10.1016/j.proci.2018.06.087. Eprint version Post-print DOI 10.1016/j.proci.2018.06.087 Publisher Elsevier BV Journal Proceedings of the Combustion Institute Rights NOTICE: this is the author’s version of a work that was accepted for publication in Proceedings of the Combustion Institute. Changes resulting from the publishing process, such as peer review, editing, corrections, structural formatting, and other quality control mechanisms may not be reflected in this document. Changes may have been made to this work since it was submitted for publication. A definitive version was subsequently published in Proceedings of the Combustion Institute, [, , (2018-07-18)] DOI: 10.1016/j.proci.2018.06.087 . © 2018. This manuscript version is made available under the CC- BY-NC-ND 4.0 license http://creativecommons.org/licenses/by- nc-nd/4.0/ Download date 27/09/2021 15:24:19 Link to Item http://hdl.handle.net/10754/630256 29 Polycyclic aromatic hydrocarbons in pyrolysis of gasoline 30 surrogates (n-heptane/iso-octane/toluene) 31 Can Shaoa, Haoyi Wanga, Nour Atefa, Zhandong Wanga*, Bingjie Chena, Maram 32 Almalkia, Yan Zhangb, Chuangchuang Caob, Jiuzhong Yangb, S.