(PAH) from G3MP2B3 Calculations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reinforcement of Styrene Butadiene Rubber Employing Poly(Isobornyl Methacrylate) (PIBOMA) As High Tg Thermoplastic Polymer
polymers Article Reinforcement of Styrene Butadiene Rubber Employing Poly(isobornyl methacrylate) (PIBOMA) as High Tg Thermoplastic Polymer Abdullah Gunaydin 1,2, Clément Mugemana 1 , Patrick Grysan 1, Carlos Eloy Federico 1 , Reiner Dieden 1 , Daniel F. Schmidt 1, Stephan Westermann 1, Marc Weydert 3 and Alexander S. Shaplov 1,* 1 Luxembourg Institute of Science and Technology (LIST), 5 Avenue des Hauts-Fourneaux, L-4362 Esch-sur-Alzette, Luxembourg; [email protected] (A.G.); [email protected] (C.M.); [email protected] (P.G.); [email protected] (C.E.F.); [email protected] (R.D.); [email protected] (D.F.S.); [email protected] (S.W.) 2 Department of Physics and Materials Science, University of Luxembourg, 2 Avenue de l’Université, L-4365 Esch-sur-Alzette, Luxembourg 3 Goodyear Innovation Center Luxembourg, L-7750 Colmar-Berg, Luxembourg; [email protected] * Correspondence: [email protected]; Tel.: +352-2758884579 Abstract: A set of poly(isobornyl methacrylate)s (PIBOMA) having molar mass in the range of 26,000–283,000 g mol−1 was prepared either via RAFT process or using free radical polymerization. ◦ These linear polymers demonstrated high glass transition temperatures (Tg up to 201 C) and thermal Citation: Gunaydin, A.; stability (T up to 230 ◦C). They were further applied as reinforcing agents in the preparation of the Mugemana, C.; Grysan, P.; onset Eloy Federico, C.; Dieden, R.; vulcanized rubber compositions based on poly(styrene butadiene rubber) (SBR). The influence of the Schmidt, D.F.; Westermann, S.; PIBOMA content and molar mass on the cure characteristics, rheological and mechanical properties of Weydert, M.; Shaplov, A.S. -
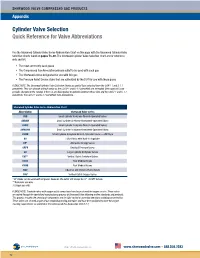
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

A Comparative Study of the Formation of Aromatics In
A COMPARATIVE STUDY OF THE FORMATION OF AROMATICS IN RICH METHANE FLAMES DOPED BY UNSATURATED COMPOUNDS H.A. GUENICHE, J. BIET, P.A. GLAUDE, R. FOURNET, F. BATTIN-LECLERC* Département de Chimie-Physique des Réactions, Nancy Université, CNRS, 1 rue Grandville, BP 20451, 54001 NANCY Cedex, France * E-mail : [email protected] ; Tel.: 33 3 83 17 51 25 , Fax : 33 3 83 37 81 20 ABSTRACT For a better modeling of the importance of the different channels leading to the first aromatic ring, we have compared the structures of laminar rich premixed methane flames doped with several unsaturated hydrocarbons: allene and propyne, because they are precursors of propargyl radicals which are well known as having an important role in forming benzene, 1,3-butadiene to put in evidence a possible production of benzene due to reactions of C4 compounds, and, finally, cyclopentene which is a source of cyclopentadienylmethylene radicals which in turn are expected to easily isomerizes to give benzene. These flames have been stabilized on a burner at a pressure of 6.7 kPa (50 Torr) using argon as dilutant, for equivalence ratios (φ) from 1.55 to 1.79. A unique mechanism, including the formation and decomposition of benzene and toluene, has been used to model the oxidation of allene, propyne, 1,3-butadiene and cyclopentene. The main reaction pathways of aromatics formation have been derived from reaction rate and sensitivity analyses and have been compared for the three types of additives. These combined analyses and comparisons can only been performed when a unique mechanism is available for all the studied additives. -

High Purity/Recovery Separation of Propylene from Propyne Using Anion Pillared Metal-Organic Framework: Application of Vacuum Swing Adsorption (VSA)
energies Article High Purity/Recovery Separation of Propylene from Propyne Using Anion Pillared Metal-Organic Framework: Application of Vacuum Swing Adsorption (VSA) Majeda Khraisheh 1, Fares AlMomani 1,* and Gavin Walker 2 1 Department of Chemical Engineering, College of Engineering, Qatar University, Doha P.O. Box 2713, Qatar; [email protected] 2 Department of Chemical Sciences, SSPC, Bernal Institute, University of Limerick, V94 T9PX Limerick, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +974-4403-4140 Abstract: Propylene is one of the world’s most important basic olefin raw material used in the produc- tion of a vast array of polymers and other chemicals. The need for high purity grade of propylene is essential and traditionally achieved by the very energy-intensive cryogenic separation. In this study, 2− a pillared inorganic anion SIF6 was used as a highly selective C3H4 due to the square grid pyrazine- based structure. Single gas adsorption revealed a very high C3H4 uptake value (3.32, 3.12, 2.97 and 2.43 mmol·g−1 at 300, 320, 340 and 360 K, respectively). The values for propylene for the same tempera- tures were 2.73, 2.64, 2.31 and 1.84 mmol·g−1, respectively. Experimental results were obtained for the two gases fitted using Langmuir and Toth models. The former had a varied degree of representation of the system with a better presentation of the adsorption of the propylene compared to the propyne system. The Toth model regression offered a better fit of the experimental data over the entire range of pressures. -

(12) United States Patent (10) Patent No.: US 8,198.491 B2 Masatoshi Et Al
USOO8198491 B2 (12) United States Patent (10) Patent No.: US 8,198.491 B2 Masatoshi et al. (45) Date of Patent: Jun. 12, 2012 (54) PROCESS FOR PREPARING 5,986,151 A 1 1/1999 Van Der Puy 2,3,3,3-TETRAFLUOROPROPENE AND $425, 23.99 Esthet al. 1,3,3,3-TETRAFLUOROPROPENE 7,833.434 B2 11/2010 Rao et al. 2005/0245773 Al 1 1/2005 Mukhopadhyay et al. (75) Inventors: Nose Masatoshi, Settsu (JP): Komatsu 2006/0258891 A1 1 1/2006 Mukhopadhyay et al. Yuzo, Settsu (JP); Sugiyama Akinari, 3.s! .k A. 239. SgO et stal al. Situ (JP); Shibanuma Takashi, Settsu 2009,0264689 A1 10, 2009 Rao et al. (JP) 2010/0200798 A1 8, 2010 Rao et al. (73) Assignee: Daikin Industries, Ltd., Osaka (JP) FOREIGN PATENT DOCUMENTS EP O 974 571 1, 2000 (*) Notice: Subject to any disclaimer, the term of this JP 63-211245 9, 1988 patent is extended or adjusted under 35 JP 11-14.0002 5, 1999 U.S.C. 154(b) by 0 days. JP 2007-320896 12/2007 WO 2008/OO2499 1, 2008 WO 2008/OO2500 1, 2008 (21) Appl. No.: 13/057,525 WO 2008/030440 3, 2008 (22) PCT Filed: Jul. 21, 2009 OTHER PUBLICATIONS International Search Report issued Mar. 3, 2010 in International (86). PCT No.: PCT/UP2O09/063314 (PCT) Application No. PCT/JP2009/063314. S371 (c)(1) PCT Written Opinion of the International Searching Authority issued (2), (4) Date:s Feb. 4, 2011 Mar.sia. 3, 2010 in International ((PCT) ) Application NoNo. PCT/JP2009/ Haszeldine et al., “Addition of FreeRadicals to Unsaturated Systems. -

Synergistic Effect of 2-Acrylamido-2-Methyl-1-Propanesulfonic Acid on the Enhanced Conductivity for Fuel Cell at Low Temperature
membranes Article Synergistic Effect of 2-Acrylamido-2-methyl- 1-propanesulfonic Acid on the Enhanced Conductivity for Fuel Cell at Low Temperature Murli Manohar * and Dukjoon Kim * School of Chemical Engineering, Sungkyunkwan University, Suwon, Kyunggi 16419, Korea * Correspondence: [email protected] (M.M.); [email protected] (D.K.) Received: 3 November 2020; Accepted: 10 December 2020; Published: 15 December 2020 Abstract: This present work focused on the aromatic polymer (poly (1,4-phenylene ether-ether-sulfone); SPEES) interconnected/ cross-linked with the aliphatic monomer (2-acrylamido -2-methyl-1-propanesulfonic; AMPS) with the sulfonic group to enhance the conductivity and make it flexible with aliphatic chain of AMPS. Surprisingly, it produced higher conductivity than that of other reported work after the chemical stability was measured. It allows optimizing the synthesis of polymer electrolyte membranes with tailor-made combinations of conductivity and stability. Membrane structure is characterized by 1H NMR and FT-IR. Weight loss of the membrane in Fenton’s reagent is not too high during the oxidative stability test. The thermal stability of the membrane is characterized by TGA and its morphology by SEM and SAXS. The prepared membranes improved 1 proton conductivity up to 0.125 Scm− which is much higher than that of Nafion N115 which is 1 0.059 Scm− . Therefore, the SPEES-AM membranes are adequate for fuel cell at 50 ◦C with reduced relative humidity (RH). Keywords: 2-acrylamido-2-methyl-1-propanesulfonic; proton-exchange membrane; conductivity; cross-linking; temperature 1. Introduction Recently, lots of polymer electrolyte membranes have been prepared from the sulfonation of aromatic polymers such as poly(arylene ether sulfone) [1–4] and modified poly(arylene ether sulfone) [5–7] for the application of electrodialysis and fuel cells as their rigid-rod backbone structures are basically quite stable in thermal and mechanical aspects. -

Cycloalkanes, Cycloalkenes, and Cycloalkynes
CYCLOALKANES, CYCLOALKENES, AND CYCLOALKYNES any important hydrocarbons, known as cycloalkanes, contain rings of carbon atoms linked together by single bonds. The simple cycloalkanes of formula (CH,), make up a particularly important homologous series in which the chemical properties change in a much more dramatic way with increasing n than do those of the acyclic hydrocarbons CH,(CH,),,-,H. The cyclo- alkanes with small rings (n = 3-6) are of special interest in exhibiting chemical properties intermediate between those of alkanes and alkenes. In this chapter we will show how this behavior can be explained in terms of angle strain and steric hindrance, concepts that have been introduced previously and will be used with increasing frequency as we proceed further. We also discuss the conformations of cycloalkanes, especially cyclo- hexane, in detail because of their importance to the chemistry of many kinds of naturally occurring organic compounds. Some attention also will be paid to polycyclic compounds, substances with more than one ring, and to cyclo- alkenes and cycloalkynes. 12-1 NOMENCLATURE AND PHYSICAL PROPERTIES OF CYCLOALKANES The IUPAC system for naming cycloalkanes and cycloalkenes was presented in some detail in Sections 3-2 and 3-3, and you may wish to review that ma- terial before proceeding further. Additional procedures are required for naming 446 12 Cycloalkanes, Cycloalkenes, and Cycloalkynes Table 12-1 Physical Properties of Alkanes and Cycloalkanes Density, Compounds Bp, "C Mp, "C diO,g ml-' propane cyclopropane butane cyclobutane pentane cyclopentane hexane cyclohexane heptane cycloheptane octane cyclooctane nonane cyclononane "At -40". bUnder pressure. polycyclic compounds, which have rings with common carbons, and these will be discussed later in this chapter. -

Recent Studies on the Aromaticity and Antiaromaticity of Planar Cyclooctatetraene
Symmetry 2010 , 2, 76-97; doi:10.3390/sym2010076 OPEN ACCESS symmetry ISSN 2073-8994 www.mdpi.com/journal/symmetry Review Recent Studies on the Aromaticity and Antiaromaticity of Planar Cyclooctatetraene Tohru Nishinaga *, Takeshi Ohmae and Masahiko Iyoda Department of Chemistry, Graduate School of Science and Engineering, Tokyo Metropolitan University, Hachioji, Tokyo 192-0397, Japan; E-Mails: [email protected] (T.O.); [email protected] (M.I.) * Author to whom correspondence should be addressed; E-Mail: [email protected]. Received: 29 December 2009; in revised form: 23 January 2010 / Accepted: 4 February 2010 / Published: 5 February 2010 Abstract: Cyclooctatetraene (COT), the first 4n π-electron system to be studied, adopts an inherently nonplanar tub-shaped geometry of D2d symmetry with alternating single and double bonds, and hence behaves as a nonaromatic polyene rather than an antiaromatic compound. Recently, however, considerable 8 π-antiaromatic paratropicity has been shown to be generated in planar COT rings even with the bond alternated D4h structure. In this review, we highlight recent theoretical and experimental studies on the antiaromaticity of hypothetical and actual planar COT. In addition, theoretically predicted triplet aromaticity and stacked aromaticity of planar COT are also briefly described. Keywords: antiaromaticity; cyclooctatetraene; NMR chemical shifts; quantum chemical calculations; ring current 1. Introduction Cyclooctatetraene (COT) was first prepared by Willstätter in 1911 [1,2]. At that time, the special stability of benzene was elusive and it was of interest to learn the reactivity of COT as the next higher vinylogue of benzene. However, unlike benzene, COT was found to be highly reactive to electrophiles just like other alkenes. -

Alkenes and Alkynes
02/21/2019 CHAPTER FOUR Alkenes and Alkynes H N O I Cl C O C O Cl F3C C Cl C Cl Efavirenz Haloprogin (antiviral, AIDS therapeutic) (antifungal, antiseptic) Chapter 4 Table of Content * Unsaturated Hydrocarbons * Introduction and hybridization * Alkenes and Alkynes * Benzene and Phenyl groups * Structure of Alkenes, cis‐trans Isomerism * Nomenclature of Alkenes and Alkynes * Configuration cis/trans, and cis/trans Isomerism * Configuration E/Z * Physical Properties of Hydrocarbons * Acid‐Base Reactions of Hydrocarbons * pka and Hybridizations 1 02/21/2019 Unsaturated Hydrocarbons • Unsaturated Hydrocarbon: A hydrocarbon that contains one or more carbon‐carbon double or triple bonds or benzene‐like rings. – Alkene: contains a carbon‐carbon double bond and has the general formula CnH2n. – Alkyne: contains a carbon‐carbon triple bond and has the general formula CnH2n‐2. Introduction Alkenes ● Hydrocarbons containing C=C ● Old name: olefins • Steroids • Hormones • Biochemical regulators 2 02/21/2019 • Alkynes – Hydrocarbons containing C≡C – Common name: acetylenes Unsaturated Hydrocarbons • Arene: benzene and its derivatives (Ch 9) 3 02/21/2019 Benzene and Phenyl Groups • We do not study benzene and its derivatives until Chapter 9. – However, we show structural formulas of compounds containing a phenyl group before that time. – The phenyl group is not reactive under any of the conditions we describe in chapters 5‐8. Structure of Alkenes • The two carbon atoms of a double bond and the four atoms bonded to them lie in a plane, with bond angles of approximately 120°. 4 02/21/2019 Structure of Alkenes • Figure 4.1 According to the orbital overlap model, a double bond consists of one bond formed by overlap of sp2 hybrid orbitals and one bond formed by overlap of parallel 2p orbitals. -

Quantification of Polycyclic Aromatic Hydrocarbon in Standard
1 Polystyrene plastic: a source and sink for polycyclic aromatic hydrocarbons in the marine 2 environment 3 Chelsea M. Rochman1§, Carlos Manzano2§, Brian T. Hentschel1, Staci L. Massey Simonich2,3, 4 Eunha Hoh4* 5 6 1Department of Biology and Coastal and Marine Institute, San Diego State University, San 7 Diego, CA 8 2Department of Chemistry, Oregon State University, Corvallis, OR 9 3Department of Environmental and Molecular Toxicology, Oregon State University, 10 Corvallis, OR 11 4Graduate School of Public Health, San Diego State University, San Diego, CA 12 13 *Corresponding Author ([email protected]) 14 §Manzano and Rochman are equal contributors to the manuscript. 15 The current address for Chelsea Rochman is the Aquatic Health Program, University of 16 California, Davis, CA and for Carlos Manzano is the Canada Centre for Inland Waters, 17 Burlington, Ontario, Canada 18 Abstract 19 Polycyclic aromatic hydrocarbons (PAHs) on virgin polystyrene (PS) and PS marine 20 debris led us to examine PS as a source and sink for PAHs in the marine environment. At two 21 locations in San Diego Bay, we measured sorption of PAHs to PS pellets, sampling at 0, 1, 3, 6, 22 9 and 12 months. We detected 25 PAHs using a new analytical method with comprehensive two- 23 dimensional gas chromatography coupled to time-of-flight mass spectrometry. Several congeners 1 24 were detected on samples before deployment. After deployment, some concentrations decreased 25 (1,3-dimethylnaphthalene and 2,6-methylnaphthalene) while most increased (2-methylanthracene 26 and all parent PAHs (PPAHs) except fluorene and fluoranthene), suggesting PS debris is a 27 source and sink for PAHs. -

Identification of a Simplest Hypervalent Hydrogen Fluoride
www.nature.com/scientificreports OPEN Identification of a Simplest Hypervalent Hydrogen Fluoride Anion in Solid Argon Received: 19 December 2016 Meng-Chen Liu1, Hui-Fen Chen2, Chih-Hao Chin1, Tzu-Ping Huang1, Yu-Jung Chen3 & Yu-Jong Accepted: 18 April 2017 Wu1,4 Published: xx xx xxxx Hypervalent molecules are one of the exceptions to the octet rule. Bonding in most hypervalent molecules is well rationalized by the Rundle–Pimentel model (three-center four-electron bond), and high ionic bonding between the ligands and the central atom is essential for stabilizing hypervalent molecules. Here, we produced one of the simplest hypervalent anions, HF−, which is known to deviate from the Rundle–Pimentel model, and identified its ro-vibrational features. High-levelab inito calculations reveal that its bond dissociation energy is comparable to that of dihalides, as supported by secondary photolysis experiments with irradiation at various wavelengths. The charge distribution analysis suggested that the F atom of HF− is negative and hypervalent and the bonding is more covalent than ionic. The octet rule indicates that atoms of the main group elements tend to gain or lose electrons in order to have eight electrons in their valence shells1, 2, similar to the electronic configuration of the noble gases. Therefore, to attain this fully filled electronic configuration, atoms combine to form molecules by sharing their valence electrons to form chemical bonds. This rule is especially applicable to the period 2 and 3 elements. Most molecules are formed by following this rule and the bonding structure of molecules can be easily recognized by using Lewis electron dot diagrams. -

Hydrogen Sulfide Capture: from Absorption in Polar Liquids to Oxide
Review pubs.acs.org/CR Hydrogen Sulfide Capture: From Absorption in Polar Liquids to Oxide, Zeolite, and Metal−Organic Framework Adsorbents and Membranes Mansi S. Shah,† Michael Tsapatsis,† and J. Ilja Siepmann*,†,‡ † Department of Chemical Engineering and Materials Science, University of Minnesota, 421 Washington Avenue SE, Minneapolis, Minnesota 55455-0132, United States ‡ Department of Chemistry and Chemical Theory Center, University of Minnesota, 207 Pleasant Street SE, Minneapolis, Minnesota 55455-0431, United States ABSTRACT: Hydrogen sulfide removal is a long-standing economic and environ- mental challenge faced by the oil and gas industries. H2S separation processes using reactive and non-reactive absorption and adsorption, membranes, and cryogenic distillation are reviewed. A detailed discussion is presented on new developments in adsorbents, such as ionic liquids, metal oxides, metals, metal−organic frameworks, zeolites, carbon-based materials, and composite materials; and membrane technologies for H2S removal. This Review attempts to exhaustively compile the existing literature on sour gas sweetening and to identify promising areas for future developments in the field. CONTENTS 4.1. Polymeric Membranes 9785 4.2. Membranes for Gas−Liquid Contact 9786 1. Introduction 9755 4.3. Ceramic Membranes 9789 2. Absorption 9758 4.4. Carbon-Based Membranes 9789 2.1. Alkanolamines 9758 4.5. Composite Membranes 9790 2.2. Methanol 9758 N 5. Cryogenic Distillation 9790 2.3. -Methyl-2-pyrrolidone 9758 6. Outlook and Perspectives 9791 2.4. Poly(ethylene glycol) Dimethyl Ether 9759 Author Information 9792 2.5. Sulfolane and Diisopropanolamine 9759 Corresponding Author 9792 2.6. Ionic Liquids 9759 ORCID 9792 3. Adsorption 9762 Notes 9792 3.1.