Transcriptome Sequencing for SNP Discovery Across Cucumis Melo
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Suggested Cultural Practices for Bitter Gourd Narinder P.S
World Vegetable Center Publication Number: 17-818 International Cooperators’ Guide Suggested Cultural Practices for Bitter Gourd Narinder P.S. Dhillon, Peter Hanson, Wallace Chen, R. Srinivasan, Lawrence Kenyon, Ray-yu Yang, Jen Wen Luoh, Maureen Mecozzi Introduction Taiwan lowlands. Growers may need to modify the practices to suit local soil, weather, pest, and Bitter gourd (Momordica charantia L.) is one disease conditions of the most popular vegetables in Southeast 1 Asia. It is a member of the cucurbit family Climate and soil requirements along with cucumber, squash, watermelon, and Bitter gourd grows well in mean air muskmelon. It provides essential micronutrients temperatures of 24-27 °C and planted in a well such as vitamin A (green variety), vitamin C, drained sandy loam or clay loam soil rich in folate calcium and dietary fiber required for organic matter. Optimum soil pH is 6.0-6.7. It good health. Bitter gourd has been used in is normally grown as an annual crop, but can traditional medicine for managing diabetes and perform as a perennial in areas with mild, frost- other diseases. In the past decade, scientific free winters. The plant thrives in the tropics evidence increasingly has shown that bitter and subtropics from lowland areas to altitudes gourd can contribute to lowering high blood of up to 1,000 m. It is more tolerant to low sugar and high blood pressure, and help in temperatures compared to other gourds, but maintaining a healthy weight. Native to China or cool temperatures will retard growth and frost India, the fast-growing vine is grown throughout will kill the plant. -

Studies on Genetic Diversity of Japanese and Vietnamese Melon Germplasm by Using Molecular Markers
Studies on genetic diversity of Japanese and Vietnamese melon germplasm by using molecular markers 2019, September Tran Phuong Dung Graduate School of Environmental and Life Science (Doctor’s course) OKAYAMA UNIVERSITY 1 Table of contents Chapter 1. General introduction .................................................................................................................. 3 1.1. Phylogenetic relationships in genus Cucumis .............................................................................. 4 1.2. Intraspecific classification and domestication history of melon ..................................................... 9 1.3. Asia – the origin center of modern melon cultivars ....................................................................... 16 Chapter 2. Genetic diversity of Japanese melon breeding lines ............................................................... 18 2.1. Introduction ..................................................................................................................................... 18 2.2. Materials and Methods ................................................................................................................... 19 2.3. Result ............................................................................................................................................... 23 2.4. Discussion ........................................................................................................................................ 28 Chapter 3. Development of RAPD‐derived STS -

Dietary Guidelines for Americans 2005
Dietary Guidelines for Americans 2005 U.S. Department of Health and Human Services U.S. Department of Agriculture www.healthierus.gov/dietaryguidelines i MESSAGE FROM THE SECRETARIES We are pleased to present the 2005 Dietary Guidelines for Americans. This document is intended to be a primary source of dietary health information for policymakers, nutrition educators, and health providers. Based on the latest scientific evidence, the 2005 Dietary Guidelines provides information and advice for choosing a nutritious diet, maintaining a healthy weight, achieving adequate exercise, and “keeping foods safe” to avoid foodborne illness. This document is based on the recommendations put forward by the Dietary Guidelines Advisory Committee. The Committee was composed of scientific experts who were responsible for reviewing and analyzing the most current dietary and nutritional information and incorporating this into a scientific evidence-based report. We want to thank them and the other public and private professionals who assisted in developing this document for their hard work and dedication. The more we learn about nutrition and exercise, the more we recognize their importance in everyday life. Children need a healthy diet for normal growth and development, and Americans of all ages may reduce their risk of chronic disease by adopting a nutritious diet and engaging in regular physical activity. However, putting this knowledge into practice is difficult. More than 90 million Americans are affected by chronic diseases and conditions that compromise their quality of life and well-being. Overweight and obesity, which are risk factors for diabetes and other chronic diseases, are more common than ever before. To correct this problem, many Americans must make significant changes in their eating habits and lifestyles. -

Formulação De Gordura Interesterificada Sem
Ana Claudia Berenhauser POTENCIAL DE TECNOLOGIAS EMERGENTES PARA A CONSERVAÇÃO E AUMENTO DO VALOR NUTRICIONAL DE LEITE HUMANO Tese apresentada ao Programa de Pós- Graduação em Ciência dos Alimentos, Centro de Ciências Agrárias, Universidade Federal de Santa Catarina, área de concentração Ciência dos Alimentos, como requisito parcial para a obtenção do Grau de Doutor em Ciência dos Alimentos. Orientadora: Profª. Drª Jane Mara Block. Florianópolis/SC 2017 3 Folha aprovacao Dedico este trabalho aos meus filhos, Bárbara, Raphael e Júlia, meu esposo, Marcus, e aos meus pais, Gabriel e Carmen. AGRADECIMENTOS A Deus, pelo dom da vida. Aos meus filhos, Bárbara, Raphael e Júlia, razão maior da minha existência. Por me permitirem experimentar o amor incondicional e pela oportunidade de lhes presentear com o alimento mais precioso e perfeito que possa existir: o leite materno. Ao meu esposo, Marcus, pelo apoio, carinho e compreensão, em todos os momentos da minha trajetória. Aos meus pais, por me ensinarem a viver a vida com dignidade, amor e dedicação. À professora Dra. Jane Mara Block pela oportunidade concedida e pela orientação. Ao professor Dr. José Vladimir de Oliveira, por acreditar na aplicação do tratamento com CO2 supercrítico no leite humano e pelo incentivo. Ao Prof. Dr. Juliano de Dea Lindner, pela orientação quanto aos procedimentos microbiológicos. À Profa. Dra. Elane Schwinden Prudêncio, pelas orientações quanto ao processo de concentração do leite humano, pelo apoio, incentivo e carinho em todas as horas. Ao Programa de Pós-Graduação pela oportunidade concedida e pelo apoio. Às colegas do Laboratório de Óleos e Gorduras, por estarem apoio e carinho. -

China - Peoples Republic Of
THIS REPORT CONTAINS ASSESSMENTS OF COMMODITY AND TRADE ISSUES MADE BY USDA STAFF AND NOT NECESSARILY STATEMENTS OF OFFICIAL U.S. GOVERNMENT POLICY Voluntary - Public Date: 12/15/2009 GAIN Report Number: CH9129 China - Peoples Republic of Post: Beijing Crop Seed of Melon Report Categories: FAIRS Subject Report Approved By: William Westman Prepared By: Mark Petry and Wu Bugang Report Highlights: On November 19, 2009 China notified "Crop Seed of Melon" to the WTO as G/TBT/N/CHN/699. Comments are due on January 18, 2010. This report contains an UNOFFICIAL translation of this draft measure. General Information: Summary On November 19, 2009 China notified “Crop Seeds of Melon” to the WTO as G/TBT/N/CHN699. Comments are due on January 18, 2010. The proposed date of adoption is 90 days after circulation by the WTO secretariat and date of entry into force is 6 months after adoption. China’s WTO notification describes this standard as specifying “the quality requirements, test methods and inspection rules for watermelon, sweet melon, wax gourd and cucumber seed. It applies to the production and sale of above mentioned melon seeds in China, which covering coating seeds and non-coating seeds.” This report contains an UNOFFICIAL translation of this draft standard. BEGIN TRANSLATION National Standard of the People’s Republic of China GB 16715.1 – 200X Replaces GB 4862 – 84 GB 16715.1 - 1996 Seed of Gourd and Vegetable Crops Part 1: Gourds (Draft for Approval) Foreword All technical aspects of this Part in GB 16715 are mandatory. GB 16715 Seed of Gourd and Vegetable Crops is divided into the following parts: Part 1: Gourd; Part 2: Chinese cabbage; Part 3: Solanaceous fruits; Part 4: Cole; Part 5: Leafy vegetables; …… This Part is the first part of GB 16715 and revision of GB 4862 - 84 The Seed of Chinese Hami Melon and GB 16715.1 - 1996 Seed of Gourd and Vegetable Crops - Gourd according to the relevant provisions of Seed Law of the People's Republic of China and the new development of the seed industry. -

DRIDIETARY REFERENCE INTAKES Thiamin, Riboflavin, Niacin, Vitamin
Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline http://www.nap.edu/catalog/6015.html DIETARY REFERENCE INTAKES DRI FOR Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline A Report of the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline and Subcommittee on Upper Reference Levels of Nutrients Food and Nutrition Board Institute of Medicine NATIONAL ACADEMY PRESS Washington, D.C. Copyright © National Academy of Sciences. All rights reserved. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline http://www.nap.edu/catalog/6015.html NATIONAL ACADEMY PRESS • 2101 Constitution Avenue, N.W. • Washington, DC 20418 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Institute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. This project was funded by the U.S. Department of Health and Human Services Office of Disease Prevention and Health Promotion, Contract No. 282-96-0033, T01; the National Institutes of Health Office of Nutrition Supplements, Contract No. N01-OD-4-2139, T024, the Centers for Disease Control and Prevention, National Center for Chronic Disease Preven- tion and Health Promotion, Division of Nutrition and Physical Activity; Health Canada; the Institute of Medicine; and the Dietary Reference Intakes Corporate Donors’ Fund. -

Molecular Analysis of the Genetic Diversity of Chinese Hami Melon and Its Relationship to the Melon Germplasm from Central and South Asia
J. Japan. Soc. Hort. Sci. 80 (1): 52–65. 2011. Available online at www.jstage.jst.go.jp/browse/jjshs1 JSHS © 2011 Molecular Analysis of the Genetic Diversity of Chinese Hami Melon and Its Relationship to the Melon Germplasm from Central and South Asia Yasheng Aierken1,2, Yukari Akashi1, Phan Thi Phuong Nhi1, Yikeremu Halidan1, Katsunori Tanaka3, Bo Long4, Hidetaka Nishida1, Chunlin Long4, Min Zhu Wu2 and Kenji Kato1* 1Graduate School of Natural Science and Technology, Okayama University, Okayama 700-8530, Japan 2Hami Melon Research Center, Xinjiang Academy of Agricultural Science, Urumuqi 830000, China 3Research Institute for Humanity and Nature, Kyoto 603-8047, Japan 4Kunming Institute of Botany, CAS, Heilongtan, Kunming, Yunnan 650204, China Chinese Hami melon consists of the varieties cassaba, chandalak, ameri, and zard. To show their genetic diversity, 120 melon accessions, including 24 accessions of Hami melon, were analyzed using molecular markers of nuclear and cytoplasmic genomes. All Hami melon accessions were classified as the large-seed type with seed length longer than 9 mm, like US and Spanish Inodorus melon. Conomon accessions grown in east China were all the small- seed type. Both large- and small-seed types were in landraces from Iran, Afghanistan, Pakistan, and Central Asia. Analysis of an SNP in the PS-ID region (Rpl16-Rpl14) and size polymorphism of ccSSR7 showed that the melon accessions consisted of three chloroplast genome types, that is, maternal lineages. Hami melon accessions were T/338 bp type, which differed from Spanish melon and US Honey Dew (T/333 bp type), indicating a different maternal lineage within group Inodorus. -

Curcumin-Based Photodynamic Sterilization for Preservation of Fresh-Cut Hami Melon
molecules Article Curcumin-Based Photodynamic Sterilization for Preservation of Fresh-Cut Hami Melon 1, 1, 1 2 1 1 Yilin Lin y , Jiamiao Hu y , Shiyang Li , Siti Sarah Hamzah , Huiqin Jiang , Arong Zhou , Shaoxiao Zeng 1 and Shaoling Lin 1,* 1 College of Food Science, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China 2 Institute for Medical Research, Jalan Pahang, 50588 Kuala Lumpur, Malaysia * Correspondence: [email protected]; Tel.: +86-15606025198 These authors contributed equally to this work. y Academic Editors: Derek J. McPhee and Francisco J. Barba Received: 24 May 2019; Accepted: 25 June 2019; Published: 27 June 2019 Abstract: Fresh-cut fruits and vegetables are the main sources of foodborne illness outbreaks with implicated pathogens such as Escherichia coli O157:H7, Salmonella, and Listeria monocytogenes. This study aimed at investigating the influence of two key parameters (concentration of curcumin and illumination time) on the effects of curcumin-based photodynamic sterilization on the preservation of fresh-cut Hami melons. The results indicated that illumination with 50 µmol/L curcumin for 60 min using a blue LED lamp reduced the total aerobic microorganism count by ~1.8 log CFU/g in fresh-cut Hami melons. Besides this, the effects of photodynamic sterilization on the soluble solids content, color, water content, firmness, and sensory indices of the fresh-cut Hami melons were also evaluated. Compared to the control group, photodynamic sterilization can effectively delay the browning rate and maintain the luminosity, firmness, water content, and soluble solids content of fresh-cut Hami melon. The sensory quality was indeed preserved well after 9 days of storage in a fridge. -
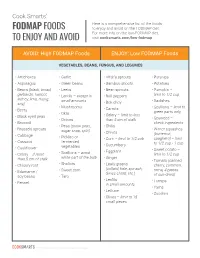
Fodmapfoods to Enjoy and Avoid
Cook Smarts’ Here is a comprehensive list of the foods FODMAP FOODS to enjoy and avoid on the FODMAP diet. For more info on the low-FODMAP diet, TO ENJOY AND AVOID visit cooksmarts.com/low-fodmap AVOID: High FODMAP Foods ENJOY: Low FODMAP Foods VEGETABLES, BEANS, FUNGUS, AND LEGUMES • Artichokes • Garlic • Alfalfa sprouts • Parsnips • Asparagus • Green beans • Bamboo shoots • Potatoes • Beans (black, broad, • Leeks • Bean sprouts • Pumpkin – garbanzo, haricot, • Lentils – except in • Bell peppers limit to 1/2 cup kidney, lima, mung, small amounts • Radishes soy) • Bok choy • Mushrooms • Scallions – limit to • Beets • Carrots • Okra green parts only • Black-eyed peas • Celery – limit to less • Onions than 5 cm of stalk • Seaweed – • Broccoli check ingredients • Peas (snow peas, • Chilis • Brussels sprouts • Winter squashes sugar snap, split) • Chives • Cabbage (butternut, • Pickled or • Corn – limit to 1/2 cob spaghetti) – limit • Cassava fermented • Cucumbers to 1/2 cup - 1 cup • Cauliflower vegetables • Eggplant • Sweet potato – • Celery – if more • Scallions – avoid limit to 1/2 cup white part of the bulb • Ginger than 5 cm of stalk • Tomato (canned, • Chicory root • Shallots • Leafy greens cherry, common, • Sweet corn (collard, kale, spinach, roma, 4 pieces • Edamame / Swiss chard, etc.) soy beans • Taro of sun-dried) • Lentils – • Turnips • Fennel in small amounts • Yams • Lettuce • Zucchini • Olives – limit to 15 small pieces © 2018 Cook Smarts | All Rights Reserved | Page 1 AVOID: High FODMAP Foods ENJOY: Low FODMAP Foods FRUITS -

02001554.Pdf
Preliminary characterization of germplasm collected in Apulia and Albania Ricciardi L., Filippetti A. in Ricciardi L. (ed.), Myrta A. (ed.), De Castro F. (ed.). Italo-Albanian cooperation for the enhancement of plant biodiversity Bari : CIHEAM Options Méditerranéennes : Série A. Séminaires Méditerranéens; n. 47 2001 pages 151-169 Article available on line / Article disponible en ligne à l’adresse : -------------------------------------------------------------------------------------------------------------------------------------------------------------------------- http://om.ciheam.org/article.php?IDPDF=2001554 -------------------------------------------------------------------------------------------------------------------------------------------------------------------------- To cite this article / Pour citer cet article -------------------------------------------------------------------------------------------------------------------------------------------------------------------------- Ricciardi L., Filippetti A. Preliminary characterization of germplasm collected in Apulia and Albania. In : Ricciardi L. (ed.), Myrta A. (ed.), De Castro F. (ed.). Italo-Albanian cooperation for the enhancement of plant biodiversity. Bari : CIHEAM, 2001. p. 151-169 (Options Méditerranéennes : Série A. Séminaires Méditerranéens; n. 47) -------------------------------------------------------------------------------------------------------------------------------------------------------------------------- http://www.ciheam.org/ http://om.ciheam.org/ -

Cucurbit Seed Production
CUCURBIT SEED PRODUCTION An organic seed production manual for seed growers in the Mid-Atlantic and Southern U.S. Copyright © 2005 by Jeffrey H. McCormack, Ph.D. Some rights reserved. See page 36 for distribution and licensing information. For updates and additional resources, visit www.savingourseeds.org For comments or suggestions contact: [email protected] For distribution information please contact: Cricket Rakita Jeff McCormack Carolina Farm Stewardship Association or Garden Medicinals and Culinaries www.carolinafarmstewards.org www.gardenmedicinals.com www.savingourseed.org www.savingourseeds.org P.O. Box 448, Pittsboro, NC 27312 P.O. Box 320, Earlysville, VA 22936 (919) 542-2402 (434) 964-9113 Funding for this project was provided by USDA-CREES (Cooperative State Research, Education, and Extension Service) through Southern SARE (Sustainable Agriculture Research and Education). Copyright © 2005 by Jeff McCormack 1 Version 1.4 November 2, 2005 Cucurbit Seed Production TABLE OF CONTENTS Scope of this manual .............................................................................................. 2 Botanical classification of cucurbits .................................................................... 3 Squash ......................................................................................................................... 4 Cucumber ................................................................................................................... 15 Melon (Muskmelon) ................................................................................................. -

Supercritical CO2 Processing of a Functional Beverage Containing Apple Juice and Aqueous Extract of Pfaffia Glomerata Roots
molecules Article Supercritical CO2 Processing of a Functional Beverage Containing Apple Juice and Aqueous Extract of Pfaffia glomerata Roots: Fructooligosaccharides Chemical Stability after Non-Thermal and Thermal Treatments Eric Keven Silva 1 , Matheus A. Bargas 1, Henrique S. Arruda 2 , Renata Vardanega 1 , Glaucia M. Pastore 2 and M. Angela A. Meireles 1,* 1 LASEFI, Department of Food Engineering, School of Food Engineering, University of Campinas, Campinas 13083-862, Brazil; [email protected] (E.K.S.); [email protected] (M.A.B.); [email protected] (R.V.) 2 Bioflavors and Bioactive Compounds Laboratory, Department of Food Science, School of Food Engineering, University of Campinas, Campinas 13083-862, Brazil; [email protected] (H.S.A.); [email protected] (G.M.P.) * Correspondence: maameireles@lasefi.com; Tel.: +55-19-98184-1414; Fax: +55-19-3521-4027 Academic Editors: Stefano Cardea and Gregory Chatel Received: 6 July 2020; Accepted: 24 August 2020; Published: 27 August 2020 Abstract: The effects of supercritical CO2 processing on the chemical stability of fructooligosaccharides (FOS) and other functional and nutritional compounds were evaluated employing non-thermal and thermal approaches. Apple juice was enriched with Pfaffia glomerata roots aqueous extract due to its high content of short-chain FOS and then subjected to different levels of temperature (40 and 60 ◦C), pressure (8 and 21 MPa), and CO2 volume ratio (20 and 50%). The percentage of CO2 volume was evaluated concerning the total volume of the high-pressure reactor. Also, the functional beverage was thermally treated at 105 ◦C for 10 min. Physicochemical properties (pH and soluble solid content), beta-ecdysone, sugars (glucose, fructose, and sucrose), and FOS (1-kestose, nystose, and fructofuranosylnystose) content were determined.