LETTER Doi:10.1038/Nature14659
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2015 White Paper Smart Learning Environments in China.Pdf
September 2015, Beijing Smart Learning Institute of Beijing Normal University White Paper: Smart Learning Environments in China 2015 (Executive Summary) Learning and Smart Learning Environments - 2 - White Paper: Smart Learning Environments in China 2015 (Executive Summary) “Livability and Innovation”: the Dual-core System of a Smart City With “People Experience of Smart Living" and "City Innovation capacity" as the dual-core, a smart city has the characteristics of smart travelling, smart living, smart learning, smart economy, smart environment and smart governance. Livability and innovation are fundamental drivers of city development, core objectives of promoting the city to operate healthily and dynamically, and efficient ways of solving those difficulties associated with the development of a "Smart City". "Smart Learning" plays a supportive role in leading city innovation capacity in culture and promoting people experience of smart living with high technology. Promoting .Entrepreneurial creativity .Internet plus economic .Convenient traffic pattern .Efficient access .Employment and Venture .Ubiquitous network access opportunities .Urban security Smart Smart .Medical and health care Economy Travelling .Civil happiness Smart Smart People Experience Environment City Innovation Living Capacity .Green building .Green energy .Green urban plan Smart Smart Governance Learning .Service policy .21st century skills .Transparency and open data .Inclusive education .Widespread use of digital government .Infusing ICT into education Leading - 3 - -

LINC00511 Exacerbated T-Cell Acute Lymphoblastic Leukemia Via Mir-195-5P/LRRK1 Axis
Bioscience Reports (2020) 40 BSR20193631 https://doi.org/10.1042/BSR20193631 Research Article LINC00511 exacerbated T-cell acute lymphoblastic leukemia via miR-195-5p/LRRK1 axis Shengli Li1, Wenwen Guo1, Huayun Geng2, Chao Wang3, Shuige Yang1 and Xinxin Xu4 1Department of Hematology, Jining No.1 People’s Hospital, No. 6 Health Road, Rencheng District, Jining 272100, Shandong, China; 2Department of Hematology, Dongchangfu People’s Hospital of Liaocheng, 281 Dongguan Street, Dongchangfu District, Liaocheng 252000, Shandong, China; 3Department of Emergency, Zi Bo Central Hospital, 54 Communist Youth League West Road, Zhangdian District, Zibo 255000, Shandong, China; 4Department of Hematology, Zi Bo Central Hospital, 54 Communist Youth League West Downloaded from http://portlandpress.com/bioscirep/article-pdf/40/5/BSR20193631/878098/bsr-2019-3631.pdf by guest on 23 September 2021 Road, Zhangdian District, Zibo, 255000 Shandong, China Correspondence: Xinxin Xu ([email protected]) T-cell acute lymphoblastic leukemia (T-ALL) is a malignant disease arising from the abnor- mal proliferation of T lymphocyte in marrow. Long non-coding RNAs (lncRNAs) are one kind of non-coding RNAs (ncRNAs), which were reported to modulate the initiation or progres- sion of diverse cancers. However, the role of LINC00511 in T-ALL was unknown. To figure out the function and mechanism of LINC00511 in T-ALL, a series of experiments were car- ried out. Based on the experimental results, we discovered that LINC00511 boosted cell proliferation and invasion, but hindered cell apoptosis in T-ALL cells. Besides, based on bio-informatics tool, miR-195-5p was selected for further exploration. Then, miR-195-5p was validated to bind with LINC00511. -

Identifying Intergenerational Risk Factors for ADHD Symptoms Using Polygenic Scores in the Norwegian Mother, Father and Child Cohort
medRxiv preprint doi: https://doi.org/10.1101/2021.02.16.21251737; this version posted February 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Intergenerational transmission of ADHD Identifying intergenerational risk factors for ADHD symptoms using polygenic scores in the Norwegian Mother, Father and Child Cohort Jean-Baptiste Pingault1,2*, Wikus Barkhuizen1*, Biyao Wang1, Laurie J. Hannigan3,4, Espen Moen Eilertsen6, Ole A. Andreassen7, Helga Ask5, Martin Tesli5,7, Ragna Bugge Askeland4,5, George Davey Smith4,9, Neil Davies4,8,9, Ted Reichborn-Kjennerud5, †Eivind Ystrom5,6, †Alexandra Havdahl3,5,6 *Equal first authors †Equal last authors 1 Division of Psychology and Language Sciences, University College London, London, United Kingdom 2 MRC Social, Genetic and Developmental Psychiatry Centre, Institute of Psychiatry, King’s College, London, United Kingdom 3 Nic Waals Institute, Lovisenberg Diaconal Hospital, Oslo, Norway 4 Medical Research Council Integrative Epidemiology Unit, University of Bristol, BS8 2BN, United Kingdom 5 Department of Mental Disorders, Norwegian Institute of Public Health, Oslo, Norway 6 PROMENTA Research Center, Department of Psychology, University of Oslo, Oslo, Norway 7 NORMENT Centre, Institute of Clinical Medicine, University of Oslo and Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway 8 K.G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, NTNU, Norwegian University of Science and Technology, Norway 9 Population Health Sciences, Bristol Medical School, University of Bristol, Barley House, Oakfield Grove, Bristol, BS8 2BN, United Kingdom Address correspondence to Dr. -

Common Genetic Variation and Schizophrenia Polygenic Risk
RESEARCH ARTICLE Neuropsychiatric Genetics Common Genetic Variation and Schizophrenia Polygenic Risk Influence Neurocognitive Performance in Young Adulthood Alex Hatzimanolis,1 Pallav Bhatnagar,2 Anna Moes,2 Ruihua Wang,1 Panos Roussos,3,4 Panos Bitsios,5 Costas N. Stefanis,6 Ann E. Pulver,1 Dan E. Arking,2 Nikolaos Smyrnis,6,7 Nicholas C. Stefanis,6,7 and Dimitrios Avramopoulos1,2* 1Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, Maryland 2McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 3Department of Psychiatry, Friedman Brain Institute and Department of Genetics and Genomics Science, Institute of Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, New York 4James J. Peters Veterans Affairs Medical Center, Bronx, New York, New York 5Department of Psychiatry and Behavioral Sciences, Faculty of Medicine, University of Crete, Heraklion, Greece 6University Mental Health Research Institute, Athens, Greece 7Department of Psychiatry, Eginition Hospital, University of Athens Medical School, Athens, Greece Manuscript Received: 3 December 2014; Manuscript Accepted: 29 April 2015 Neurocognitive abilities constitute complex traits with consid- erable heritability. Impaired neurocognition is typically ob- How to Cite this Article: served in schizophrenia (SZ), whereas convergent evidence Hatzimanolis A, Bhatnagar P, Moes A, has shown shared genetic determinants between neurocognition Wang R, Roussos P, Bitsios P, Stefanis CN, and SZ. Here, we report a genome-wide association study Pulver AE, Arking DE, Smyrnis N, Stefanis (GWAS) on neuropsychological and oculomotor traits, linked NC, Avramopoulos D. 2015. Common to SZ, in a general population sample of healthy young males Genetic Variation and Schizophrenia ¼ (n 1079). -

Polygenic Methods and Their Application to Psychiatric Traits
This is the peer reviewed version of the following article: Wray, N. R., & Visscher, P. M. (2015). Quantitative genetics of disease traits. Wray, N. R., Lee, S. H., Mehta, D., Vinkhuyzen, A. A., Dudbridge, F., & Middeldorp, C. M. (2014). Research review: polygenic methods and their application to psychiatric traits. Journal of Child Psychology and Psychiatry, 55(10), 1068–1087. DOI: 10.1111/jcpp.12295 . This article may be used for non-commercial purposes in accordance with Wiley Terms and Conditions for self-archiving. Polygenic methods and their application to psychiatric traits Naomi R Wray1*, S Hong Lee1, Divya Mehta1, Anna AE Vinkhuyzen1, Frank Dudbridge2, Christel M Middeldorp3,4 1. The University of Queensland, Queensland Brain Institute, Queensland, Australia 4068 2. Department of Non-Communicable Disease Epidemiology, London School of Hygiene and Tropical Medicine, London, UK 3. Department of Biological Psychology, Neuroscience Campus Amsterdam, VU University Amsterdam, The Netherlands 4. Department of Child and Adolescent Psychiatry, GGZ inGeest/VU University Medical Center, Amsterdam, The Netherlands * Corresponding author. [email protected] Abstract Background Despite evidence from twin and family studies for an important contribution of genetic factors to both childhood and adult onset psychiatric disorders, identifying robustly associated specific DNA variants has proved challenging. In the pre-genomics era the genetic architecture (number, frequency and effect size of risk variants) of complex genetic disorders was unknown. Empirical evidence for the genetic architecture of psychiatric disorders is emerging from the genetic studies of the last five years. Methods and scope We review the methods investigating the polygenic nature of complex disorders. -

The BRCA1 Tumor Suppressor
Journal of Translational Science Review Article ISSN: 2059-268X The BRCA1 tumor suppressor: potential long-range interactions of the BRCA1 promoter and the risk of breast cancer Beata Anna Bielinska* Department of Molecular and Translational Oncology, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Poland Abstract Breast cancer is a complex disease with different phenotypes associated with genetic and non-genetic risk factors. An aberrant expression of the BRCA1 tumor suppressor as well as dysfunction of BRCA1 protein caused by germline mutations are implicated in breast cancer aethiology. BRCA1 plays a crucial role in genome and epigenome stability. Its expression is auto regulated and modulated by various cellular signals including metabolic status, hypoxia, DNA damage, estrogen stimulation. The review describes breast cancer risk factors, the BRCA1 gene expression and functions as well as covers the role of long-range genomic interactions, which emerge as regulators of gene expression and moderators of genomic communication. The potential long-range interactions of the BRCA1 promoter (driven by polymorphic variant rs11655505 C/T, as an example) and their possible impact on the BRCA1 gene regulation and breast cancer risk are also discussed. Introduction efficiency of BRCA1 transcription [9]. However, to date the mechanism underlying effect of the BRCA1 promoter variant rs11655505 C>T Female breast cancer is one of the most common cancers worldwide remains unknown. with app.1.68 million cases diagnosed annually [1]. It is a complex disease characterized by molecular and phenotypic heterogeneity Besides high risk BRCA1/BRCA2 mutations, moderate risk observed both within populations and intra-individually, within mutations such as those found in DNA repair genes (CHEK2, ATM, single tumor cells in a spatiotemporal manner, as pointed out by PALB2) also demonstrate familial clustering. -

2020 Annual Report
ANNUAL REPORT 2020 Contents Definitions and Glossary of Technical Terms 2 Corporate Information 11 Chairman’s Statement 13 Financial Highlights 16 Management Discussion and Analysis 17 Corporate Governance Report 30 Profiles of Directors, Supervisors and 47 Senior Management Report of the Board of Directors 66 Environmental, Social and Governance Report 99 Independent Auditor’s Report 138 Consolidated Statement of Profit or Loss and 143 Other Comprehensive Income Consolidated Statement of Financial Position 144 Consolidated Statement of Changes in Equity 146 Consolidated Statement of Cash Flows 147 Notes to Financial Statements 149 2 2020 ANNUAL REPORT Definitions and Glossary of Technical Terms “2020 AGM” the 2020 annual general meeting of the Company to be convened and held on Thursday, 24 June 2021 or the adjournment thereof “2020 Final Dividend” the final dividend proposed by the Board to be paid to the Shareholders in the form of a cash dividend of RMB0.180 (tax inclusive) per Share "2021-2023 General Services the agreement dated 2 June 2020 and entered into between the Framework Agreement" Company and Qilu Transportation for the procurement of the general highway business operation services from Qilu Transportation and its relevant subordinated entities for the three years ending 31 December 2023 “Acquisition Circular” the circular of the Company dated 26 June 2020 in relation to, among others, the acquisition of the Deshang and Shennan Expressways Toll Collection Rights by the Company from Qilu Transportation “Administration for -

Factsheet Logistics 08082018
SIME DARBY LOGISTICSAugust 2018 Operates four ports across Weifang and Jining in Shandong,China Weifang Sime Darby Port lies within the prime region of the Bohai Sea’s economic belt, one of the busiest seaways in the world Total handling capacity of approximately 60.8 million metric tonnes (MT) per annum August 2018 SIME DARBY LOGISTICS Dongying Binzhou Denzhou Yantai Weihai Qingdao Jinan Zibo Weifang Liaocheng Taian Weifang Sime Darby Port Co Ltd Rizhao Jining Linyi Heze Zaozhuang People‘s Republic of China Jining Sime Darby Port Co, Ltd Jining Sime Darby Longgong Port Co, Ltd Jining Sime Darby Taiping Port Co,Ltd Sime Darby Logistics is involved in ports and logistics Shandong businesses in the province of Shandong, in Eastern China. The company employs approximately 1,000 employees based in Weifang and Jining. Sime Darby Logistics’ first foray into Shandong began in the prefecture-level city of Weifang with the incorporation of Weifang Sime Darby Port Co Ltd and Weifang Sime Darby Water Management Co Ltd in 2005. Today, Sime Darby Logistics’ operations in China have expanded to include three river ports in the prefecture- level city of Jining. August 2018 PORTS AND LOGISTICS Located in the prime region of the Bohai Sea’s economic Weifang Sime Darby Port Co L td belt, Weifang Port is a seaport that handles bulk (dry and Weifang Sime Darby Port Co Ltd (”Weifang Port”) liquid), general cargo and containers. Weifang Port commenced operations in 1998, following an invitation currently has a total handling capacity of 44.4 million MT from the Weifang Municipal Government to invest in the per annum with an annual throughput of 26.5 million MT. -

Within-Sibship GWAS Improve Estimates of Direct Genetic Effects
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.05.433935; this version posted March 7, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Within-sibship GWAS improve estimates of direct genetic effects Laurence J Howe1,2 *, Michel G Nivard3, Tim T Morris1,2, Ailin F Hansen4, Humaira Rasheed4, Yoonsu Cho1,2, Geetha Chittoor5, Penelope A Lind6,7,8, Teemu Palviainen9, Matthijs D van der Zee3, Rosa Cheesman10,11, Massimo Mangino12,13, Yunzhang Wang14, Shuai Li15,16,17, Lucija Klaric18, Scott M Ratliff19, Lawrence F Bielak19, Marianne Nygaard20,21, Chandra A Reynolds22, Jared V Balbona23,24, Christopher R Bauer25,26, Dorret I Boomsma3,27, Aris Baras28, Archie Campbell29, Harry Campbell30, Zhengming Chen31,32, Paraskevi Christofidou12, Christina C Dahm33, Deepika R Dokuru23,24, Luke M Evans24,34, Eco JC de Geus3,35, Sudheer Giddaluru36,37, Scott D Gordon38, K. Paige Harden39, Alexandra Havdahl40,41, W. David Hill42,43, Shona M Kerr18, Yongkang Kim24, Hyeokmoon Kweon44, Antti Latvala9,45, Liming Li46, Kuang Lin31, Pekka Martikainen47,48,49, Patrik KE Magnusson14, Melinda C Mills50, Deborah A Lawlor1,2,51, John D Overton27, Nancy L Pedersen14, David J Porteous25, Jeffrey Reid28, Karri Silventoinen47, Melissa C Southey17,52,53, Travis T Mallard39, Elliot M Tucker-Drob39, Margaret J Wright54, Social Science Genetic Association Consortium, Within Family -

Cereal Series/Protein Series Jiangxi Cowin Food Co., Ltd. Huangjindui
产品总称 委托方名称(英) 申请地址(英) Huangjindui Industrial Park, Shanggao County, Yichun City, Jiangxi Province, Cereal Series/Protein Series Jiangxi Cowin Food Co., Ltd. China Folic acid/D-calcium Pantothenate/Thiamine Mononitrate/Thiamine East of Huangdian Village (West of Tongxingfengan), Kenli Town, Kenli County, Hydrochloride/Riboflavin/Beta Alanine/Pyridoxine Xinfa Pharmaceutical Co., Ltd. Dongying City, Shandong Province, 257500, China Hydrochloride/Sucralose/Dexpanthenol LMZ Herbal Toothpaste Liuzhou LMZ Co.,Ltd. No.282 Donghuan Road,Liuzhou City,Guangxi,China Flavor/Seasoning Hubei Handyware Food Biotech Co.,Ltd. 6 Dongdi Road, Xiantao City, Hubei Province, China SODIUM CARBOXYMETHYL CELLULOSE(CMC) ANQIU EAGLE CELLULOSE CO., LTD Xinbingmaying Village, Linghe Town, Anqiu City, Weifang City, Shandong Province No. 569, Yingerle Road, Economic Development Zone, Qingyun County, Dezhou, biscuit Shandong Yingerle Hwa Tai Food Industry Co., Ltd Shandong, China (Mainland) Maltose, Malt Extract, Dry Malt Extract, Barley Extract Guangzhou Heliyuan Foodstuff Co.,LTD Mache Village, Shitan Town, Zengcheng, Guangzhou,Guangdong,China No.3, Xinxing Road, Wuqing Development Area, Tianjin Hi-tech Industrial Park, Non-Dairy Whip Topping\PREMIX Rich Bakery Products(Tianjin)Co.,Ltd. Tianjin, China. Edible oils and fats / Filling of foods/Milk Beverages TIANJIN YOSHIYOSHI FOOD CO., LTD. No. 52 Bohai Road, TEDA, Tianjin, China Solid beverage/Milk tea mate(Non dairy creamer)/Flavored 2nd phase of Diqiuhuanpo, Economic Development Zone, Deqing County, Huzhou Zhejiang Qiyiniao Biological Technology Co., Ltd. concentrated beverage/ Fruit jam/Bubble jam City, Zhejiang Province, P.R. China Solid beverage/Flavored concentrated beverage/Concentrated juice/ Hangzhou Jiahe Food Co.,Ltd No.5 Yaojia Road Gouzhuang Liangzhu Street Yuhang District Hangzhou Fruit Jam Production of Hydrolyzed Vegetable Protein Powder/Caramel Color/Red Fermented Rice Powder/Monascus Red Color/Monascus Yellow Shandong Zhonghui Biotechnology Co., Ltd. -

LDL Polygenic Risk Scores in Practice
University of Copenhagen & Copenhagen University Hospital LDL polygenic risk scores in practice Anne Tybjærg-Hansen MD DMSc Professor, Chief Physician Copenhagen University Hospital and Faculty of Health and Medical Sciences, University of Copenhagen, Denmark FH Global Summit, Atlanta 211019 Outline • Introduction to polygenic risk score • Polygenic risk scores for LDL cholesterol and FH • Polygenic risk scores for CAD and FH – polygenic versus omnigenic scores • Take home message Introduction to polygenic risk score Genome-wide association study of LDL cholesterol (from GLGC) P<10-8 Willer CJ et al. Nat Genet 2013 Creating a polygenic score GWAS SNPs Individually: tiny effects ComBined: larger effect Effect of gene score on trait level 1.0 1.2 Odds ratio Genome-wide association study of LDL cholesterol Polygenic risk score P<10-8 Willer CJ et al. Nat Genet 2013 Genome-wide association studies (GWAS’) for lipids In sufficiently large samples, many ~ 100,000 previously non-significant loci would European ancestry become nominally significant, 95 loci although effect sizes are close to zero. ~ 600,000 ~85,000 non-Europeans ~200,000 >118 novel loci 7,898 non-Europeans (~268 known loci) 62 novel loci 30.4 mill SNPs Klarin et al. 2010 2013 2018 Polygenic risk scores for LDL cholesterol and Familial Hypercholesterolemia Rigshospitalet The Centre of Diagnostic Investigations Diagnostic criteria for FH 1/220 individuals • Definite FH: > 8 points • Probable FH: 6-8 points • Possible FH: 3-5 points • Unlikely FH: 0-2 points Mette Christoffersen 9 Rigshospitalet The Centre of Diagnostic Investigations Clinical vs. mutation diagnosis Polygenic cause? 20-40% High Lp(a)? 60-80 % Polygenic cause? Modified from Eur Heart J. -
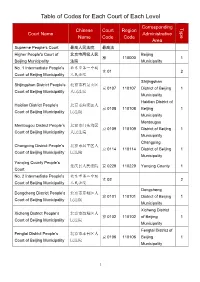
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115