Download This Publication (PDF File)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phylogeny and Classification of Cryptodiscus, with a Taxonomic Synopsis of the Swedish Species
Fungal Diversity Phylogeny and classification of Cryptodiscus, with a taxonomic synopsis of the Swedish species Baloch, E.1,3*, Gilenstam, G.2 and Wedin, M.1 1Department of Cryptogamic Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05 Stockholm, Sweden. 2Department of Ecology and Environmental Sciences, Umeå University, SE-901 87 Umeå, Sweden. 3Jodrell Laboratory, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3AB, UK. Baloch, E., Gilenstam, G. and Wedin, M. (2009). Phylogeny and classification of Cryptodiscus, with a taxonomic synopsis of the Swedish species. Fungal Diversity 38: 51-68. The phylogeny, taxonomy and classification of Cryptodiscus are examined. The current generic and species delimitations, and the relationship of the genus within the Ostropomycetidae, are tested by molecular phylogenetic analyses of the nuclear ITS and LSU rDNA and the mitochondrial SSU rDNA. In our new circumscription Cryptodiscus is a monophyletic group of saprotrophic and lichenized fungi characterized by small, urceolate apothecia, mostly hyaline ascomatal walls without any embedded crystals, no clear periphysoids, and with oblong to narrow- cylindrical septate ascospores. Cryptodiscus forms a well-supported clade together with Absconditella and the remaining Stictidaceae. Paschelkiella and Bryophagus are synonymised with Cryptodiscus. Species excluded from Cryptodiscus are Cryptodiscus anguillosporus, C. angulosus, C. microstomus, and C. rhopaloides. Cryptodiscus in Sweden is revised and six species are accepted, of which one is newly described: C. foveolaris, C. gloeocapsa comb. nov. (≡ Bryophagus gloeocapsa), C. incolor sp. nov., C. pallidus, C. pini comb. nov. (≡ Paschelkiella pini), and the rediscovered species C. tabularum. The additional new combinations Cryptodiscus similis comb. nov. and C. -

New Or Interesting Lichens and Lichenicolous Fungi from Belgium, Luxembourg and Northern France
New or interesting lichens and lichenicolous fungi from Belgium, Luxembourg and northern France. X Emmanuël SÉRUSIAUX1, Paul DIEDERICH2, Damien ERTZ3, Maarten BRAND4 & Pieter VAN DEN BOOM5 1 Plant Taxonomy and Conservation Biology Unit, University of Liège, Sart Tilman B22, B-4000 Liège, Belgique ([email protected]) 2 Musée national d’histoire naturelle, 25 rue Munster, L-2160 Luxembourg, Luxembourg ([email protected]) 3 Jardin Botanique National de Belgique, Domaine de Bouchout, B-1860 Meise, Belgium ([email protected]) 4 Klipperwerf 5, NL-2317 DX Leiden, the Netherlands ([email protected]) 5 Arafura 16, NL-5691 JA Son, the Netherlands ([email protected]) Sérusiaux, E., P. Diederich, D. Ertz, M. Brand & P. van den Boom, 2006. New or interesting lichens and lichenicolous fungi from Belgium, Luxembourg and northern France. X. Bul- letin de la Société des naturalistes luxembourgeois 107 : 63-74. Abstract. Review of recent literature and studies on large and mainly recent collections of lichens and lichenicolous fungi led to the addition of 35 taxa to the flora of Belgium, Lux- embourg and northern France: Abrothallus buellianus, Absconditella delutula, Acarospora glaucocarpa var. conspersa, Anema nummularium, Anisomeridium ranunculosporum, Artho- nia epiphyscia, A. punctella, Bacidia adastra, Brodoa atrofusca, Caloplaca britannica, Cer- cidospora macrospora, Chaenotheca laevigata, Collemopsidium foveolatum, C. sublitorale, Coppinsia minutissima, Cyphelium inquinans, Involucropyrenium squamulosum, Lecania fructigena, Lecanora conferta, L. pannonica, L. xanthostoma, Lecidea variegatula, Mica- rea micrococca, Micarea subviridescens, M. vulpinaris, Opegrapha prosodea, Parmotrema stuppeum, Placynthium stenophyllum var. isidiatum, Porpidia striata, Pyrenidium actinellum, Thelopsis rubella, Toninia physaroides, Tremella coppinsii, Tubeufia heterodermiae, Verru- caria acrotella and Vezdaea stipitata. -

Lichenicolous Biota (Nos 201–230)
ZOBODAT - www.zobodat.at Zoologisch-Botanische Datenbank/Zoological-Botanical Database Digitale Literatur/Digital Literature Zeitschrift/Journal: Fritschiana Jahr/Year: 2015 Band/Volume: 80 Autor(en)/Author(s): Hafellner Josef Artikel/Article: Lichenicolous Biota (Nos 201-230) 21-41 - 21 - Lichenicolous Biota (Nos 201–230) Josef HAFELLNER* HAFELLNER Josef 2015: Lichenicolous Biota (Nos 201–230). – Frit- schiana (Graz) 80: 21–41. - ISSN 1024-0306. Abstract: The 9th fascicle (30 numbers) of the exsiccata 'Lichenicolous Biota' is published. The issue contains ma- terial of 20 non-lichenized fungal taxa (14 teleomorphs of ascomycetes, 4 anamorphic states of ascomycetes, 2 an- amorphic states of basidiomycetes) and 9 lichenized as- comycetes, including paratype material of Dimelaena li- chenicola K.Knudsen et al. (no 223), Miriquidica invadens Hafellner et al. (no 226, 227), and Stigmidium xantho- parmeliarum Hafellner (no 210). Furthermore, collections of the type species of the following genera are distributed: Illosporiopsis (I. christiansenii), Illosporium (I. carneum), Marchandiomyces (M. corallinus), Marchandiobasidium (M. aurantiacum, sub Erythricium aurantiacum), Micro- calicium (M. disseminatum), Nigropuncta (N. rugulosa), Paralecanographa (P. grumulosa), Phaeopyxis (P. punc- tum), Placocarpus (P. schaereri), Rhagadostoma (R. li- chenicola), and Stigmidium (S. schaereri). *Institut für Pflanzenwissenschaften, NAWI Graz, Karl-Franzens-Universität, Holteigasse 6, 8010 Graz, AUSTRIA e-mail: [email protected] Introduction The exsiccata 'Lichenicolous Biota' is continued with fascicle 9, containing 30 numbers. The exsiccata covers all lichenicolous biota, i.e., it is open not only to non- lichenized and lichenized fungi, but also to myxomycetes, bacteria, and even animals, whenever they cause a characteristic symptom on their host (e.g. discoloration or galls). -
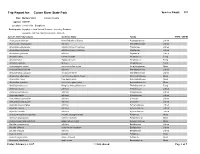
Cuivre Bryophytes
Trip Report for: Cuivre River State Park Species Count: 335 Date: Multiple Visits Lincoln County Agency: MODNR Location: Lincoln Hills - Bryophytes Participants: Bryophytes from Natural Resource Inventory Database Bryophyte List from NRIDS and Bruce Schuette Species Name (Synonym) Common Name Family COFC COFW Acarospora unknown Identified only to Genus Acarosporaceae Lichen Acrocordia megalospora a lichen Monoblastiaceae Lichen Amandinea dakotensis a button lichen (crustose) Physiaceae Lichen Amandinea polyspora a button lichen (crustose) Physiaceae Lichen Amandinea punctata a lichen Physiaceae Lichen Amanita citrina Citron Amanita Amanitaceae Fungi Amanita fulva Tawny Gresette Amanitaceae Fungi Amanita vaginata Grisette Amanitaceae Fungi Amblystegium varium common willow moss Amblystegiaceae Moss Anisomeridium biforme a lichen Monoblastiaceae Lichen Anisomeridium polypori a crustose lichen Monoblastiaceae Lichen Anomodon attenuatus common tree apron moss Anomodontaceae Moss Anomodon minor tree apron moss Anomodontaceae Moss Anomodon rostratus velvet tree apron moss Anomodontaceae Moss Armillaria tabescens Ringless Honey Mushroom Tricholomataceae Fungi Arthonia caesia a lichen Arthoniaceae Lichen Arthonia punctiformis a lichen Arthoniaceae Lichen Arthonia rubella a lichen Arthoniaceae Lichen Arthothelium spectabile a lichen Uncertain Lichen Arthothelium taediosum a lichen Uncertain Lichen Aspicilia caesiocinerea a lichen Hymeneliaceae Lichen Aspicilia cinerea a lichen Hymeneliaceae Lichen Aspicilia contorta a lichen Hymeneliaceae Lichen -

<I>Cyanodermella Asteris</I> Sp. Nov. (<I>Ostropales</I>)
MYCOTAXON ISSN (print) 0093-4666 (online) 2154-8889 Mycotaxon, Ltd. ©2017 January–March 2017—Volume 132, pp. 107–123 http://dx.doi.org/10.5248/132.107 Cyanodermella asteris sp. nov. (Ostropales) from the inflorescence axis of Aster tataricus Linda Jahn1,*, Thomas Schafhauser2, Stefan Pan2, Tilmann Weber2,7, Wolfgang Wohlleben2, David Fewer3, Kaarina Sivonen3, Liane Flor4, Karl-Heinz van Pée4, Thibault Caradec5, Philippe Jacques5,8, Mieke M.E. Huijbers6,9, Willem J.H. van Berkel6 & Jutta Ludwig-Müller1,* 1 Institut für Botanik, Technische Universität Dresden, 01062 Dresden, Germany 2 Mikrobiologie und Biotechnologie, Interfakultäres Institut für Mikrobiologie und Infektionsmedizin, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 28, 72076 Tübingen, Germany 3 Microbiology and Biotechnology Division, Dept. of Food and Environmental Sciences, University of Helsinki, Viikinkaari 9, FIN-00014, Helsinki, Finland 4 Allgemeine Biochemie, Technische Universität Dresden, 01069 Dresden, Germany 5 Laboratoire ProBioGEM, Université Lille1- Sciences et Technologies, Villeneuve d’Ascq, France 6 Laboratory of Biochemistry, Wageningen University, Dreijenlaan 3, 6703 HA Wageningen, The Netherlands 7 moved to: Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kemitorvet Bygning 220, 2800 Kgs. Lyngby, Denmark 8 moved to: Gembloux Agro-Bio Tech, Université de Liege, Passage des Déportés 2, 5030 Gembloux, Belgium 9 moved to: Department of Biotechnology, Technical University Delft, Van der Maasweg 9, 2629 HZ Delft, The Netherlands *Correspondence to: [email protected], [email protected] Abstract—An endophytic fungus isolated from the inflorescence axis ofAster tataricus is proposed as a new species. Phylogenetic analyses based on sequences from the ribosomal DNA cluster (the ITS1+5.8S+ITS2, 18S, and 28S regions) and the RPB2 gene revealed a relationship between the unknown fungus and the Stictidaceae lineage of the Ostropales. -

Molecular Phylogenetic Study at the Generic Boundary Between the Lichen-Forming Fungi Caloplaca and Xanthoria (Ascomycota, Teloschistaceae)
Mycol. Res. 107 (11): 1266–1276 (November 2003). f The British Mycological Society 1266 DOI: 10.1017/S0953756203008529 Printed in the United Kingdom. Molecular phylogenetic study at the generic boundary between the lichen-forming fungi Caloplaca and Xanthoria (Ascomycota, Teloschistaceae) Ulrik SØCHTING1 and Franc¸ ois LUTZONI2 1 Department of Mycology, Botanical Institute, University of Copenhagen, O. Farimagsgade 2D, DK-1353 Copenhagen K, Denmark. 2 Department of Biology, Duke University, Durham, NC 27708-0338, USA. E-mail : [email protected] Received 5 December 2001; accepted 5 August 2003. A molecular phylogenetic analysis of rDNA was performed for seven Caloplaca, seven Xanthoria, one Fulgensia and five outgroup species. Phylogenetic hypotheses are constructed based on nuclear small and large subunit rDNA, separately and in combination. Three strongly supported major monophyletic groups were revealed within the Teloschistaceae. One group represents the Xanthoria fallax-group. The second group includes three subgroups: (1) X. parietina and X. elegans; (2) basal placodioid Caloplaca species followed by speciations leading to X. polycarpa and X. candelaria; and (3) a mixture of placodioid and endolithic Caloplaca species. The third main monophyletic group represents a heterogeneous assemblage of Caloplaca and Fulgensia species with a drastically different metabolite content. We report here that the two genera Caloplaca and Xanthoria, as well as the subgenus Gasparrinia, are all polyphyletic. The taxonomic significance of thallus morphology in Teloschistaceae and the current delimitation of the genus Xanthoria is discussed in light of these results. INTRODUCTION Taxonomy of Teloschistaceae and its genera The Teloschistaceae is a well-delimited family of Hawksworth & Eriksson (1986) assigned the Teloschis- lichenized fungi. -

Insights Into the Ecology and Genetics of Lichens with a Cyanobacterial Photobiont
Insights into the Ecology and Genetics of Lichens with a Cyanobacterial Photobiont Katja Fedrowitz Faculty of Natural Resources and Agricultural Sciences Department of Ecology Uppsala Doctoral Thesis Swedish University of Agricultural Sciences Uppsala 2011 Acta Universitatis agriculturae Sueciae 2011:96 Cover: Lobaria pulmonaria, Nephroma bellum, and fallen bark in an old-growth forest in Finland with Populus tremula. Part of the tRNALeu (UAA) sequence in an alignment. (photos: K. Fedrowitz) ISSN 1652-6880 ISBN 978-91-576-7640-5 © 2011 Katja Fedrowitz, Uppsala Print: SLU Service/Repro, Uppsala 2011 Insights into the Ecology and Genetics of Lichens with a Cyanobacterial Photobiont Abstract Nature conservation requires an in-depth understanding of the ecological processes that influence species persistence in the different phases of a species life. In lichens, these phases comprise dispersal, establishment, and growth. This thesis aimed at increasing the knowledge on epiphytic cyanolichens by studying different aspects linked to these life stages, including species colonization extinction dynamics, survival and vitality of lichen transplants, and the genetic symbiont diversity in the genus Nephroma. Paper I reveals that local colonizations, stochastic, and deterministic extinctions occur in several epiphytic macrolichens. Species habitat-tracking metapopulation dynamics could partly be explained by habitat quality and size, spatial connectivity, and possibly facilitation by photobiont sharing. Simulations of species future persistence suggest stand-level extinction risk for some infrequent sexually dispersed species, especially when assuming low tree numbers and observed tree fall rates. Forestry practices influence the natural occurrence of species, and retention of trees at logging is one measure to maintain biodiversity. However, their long-term benefit for biodiversity is still discussed. -

Umbilicariaceae Phylogeny TAXON 66 (6) • December 2017: 1282–1303
Davydov & al. • Umbilicariaceae phylogeny TAXON 66 (6) • December 2017: 1282–1303 Umbilicariaceae (lichenized Ascomycota) – Trait evolution and a new generic concept Evgeny A. Davydov,1 Derek Peršoh2 & Gerhard Rambold3 1 Altai State University, Lenin Ave. 61, Barnaul, 656049 Russia 2 Ruhr-Universität Bochum, AG Geobotanik, Gebäude ND 03/170, Universitätsstraße 150, 44801 Bochum, Germany 3 University of Bayreuth, Plant Systematics, Mycology Dept., Universitätsstraße 30, NW I, 95445 Bayreuth, Germany Author for correspondence: Evgeny A. Davydov, [email protected] ORCID EAD, http://orcid.org/0000-0002-2316-8506; DP, http://orcid.org/0000-0001-5561-0189 DOI https://doi.org/10.12705/666.2 Abstract To reconstruct hypotheses on the evolution of Umbilicariaceae, 644 sequences from three independent DNA regions were used, 433 of which were newly produced. The study includes a representative fraction (presumably about 80%) of the known species diversity of the Umbilicariaceae s.str. and is based on the phylograms obtained using maximum likelihood and a Bayesian phylogenetic inference framework. The analyses resulted in the recognition of eight well-supported clades, delimited by a combination of morphological and chemical features. None of the previous classifications within Umbilicariaceae s.str. were supported by the phylogenetic analyses. The distribution of the diagnostic morphological and chemical traits against the molecular phylogenetic topology revealed the following patterns of evolution: (1) Rhizinomorphs were gained at least four times independently and are lacking in most clades grouping in the proximity of Lasallia. (2) Asexual reproductive structures, i.e., thalloconidia and lichenized dispersal units, appear more or less mutually exclusive, being restricted to different clades. -

An Evolving Phylogenetically Based Taxonomy of Lichens and Allied Fungi
Opuscula Philolichenum, 11: 4-10. 2012. *pdf available online 3January2012 via (http://sweetgum.nybg.org/philolichenum/) An evolving phylogenetically based taxonomy of lichens and allied fungi 1 BRENDAN P. HODKINSON ABSTRACT. – A taxonomic scheme for lichens and allied fungi that synthesizes scientific knowledge from a variety of sources is presented. The system put forth here is intended both (1) to provide a skeletal outline of the lichens and allied fungi that can be used as a provisional filing and databasing scheme by lichen herbarium/data managers and (2) to announce the online presence of an official taxonomy that will define the scope of the newly formed International Committee for the Nomenclature of Lichens and Allied Fungi (ICNLAF). The online version of the taxonomy presented here will continue to evolve along with our understanding of the organisms. Additionally, the subfamily Fissurinoideae Rivas Plata, Lücking and Lumbsch is elevated to the rank of family as Fissurinaceae. KEYWORDS. – higher-level taxonomy, lichen-forming fungi, lichenized fungi, phylogeny INTRODUCTION Traditionally, lichen herbaria have been arranged alphabetically, a scheme that stands in stark contrast to the phylogenetic scheme used by nearly all vascular plant herbaria. The justification typically given for this practice is that lichen taxonomy is too unstable to establish a reasonable system of classification. However, recent leaps forward in our understanding of the higher-level classification of fungi, driven primarily by the NSF-funded Assembling the Fungal Tree of Life (AFToL) project (Lutzoni et al. 2004), have caused the taxonomy of lichen-forming and allied fungi to increase significantly in stability. This is especially true within the class Lecanoromycetes, the main group of lichen-forming fungi (Miadlikowska et al. -

Lichen Functional Trait Variation Along an East-West Climatic Gradient in Oregon and Among Habitats in Katmai National Park, Alaska
AN ABSTRACT OF THE THESIS OF Kaleigh Spickerman for the degree of Master of Science in Botany and Plant Pathology presented on June 11, 2015 Title: Lichen Functional Trait Variation Along an East-West Climatic Gradient in Oregon and Among Habitats in Katmai National Park, Alaska Abstract approved: ______________________________________________________ Bruce McCune Functional traits of vascular plants have been an important component of ecological studies for a number of years; however, in more recent times vascular plant ecologists have begun to formalize a set of key traits and universal system of trait measurement. Many recent studies hypothesize global generality of trait patterns, which would allow for comparison among ecosystems and biomes and provide a foundation for general rules and theories, the so-called “Holy Grail” of ecology. However, the majority of these studies focus on functional trait patterns of vascular plants, with a minority examining the patterns of cryptograms such as lichens. Lichens are an important component of many ecosystems due to their contributions to biodiversity and their key ecosystem services, such as contributions to mineral and hydrological cycles and ecosystem food webs. Lichens are also of special interest because of their reliance on atmospheric deposition for nutrients and water, which makes them particularly sensitive to air pollution. Therefore, they are often used as bioindicators of air pollution, climate change, and general ecosystem health. This thesis examines the functional trait patterns of lichens in two contrasting regions with fundamentally different kinds of data. To better understand the patterns of lichen functional traits, we examined reproductive, morphological, and chemical trait variation along precipitation and temperature gradients in Oregon. -

H. Thorsten Lumbsch VP, Science & Education the Field Museum 1400
H. Thorsten Lumbsch VP, Science & Education The Field Museum 1400 S. Lake Shore Drive Chicago, Illinois 60605 USA Tel: 1-312-665-7881 E-mail: [email protected] Research interests Evolution and Systematics of Fungi Biogeography and Diversification Rates of Fungi Species delimitation Diversity of lichen-forming fungi Professional Experience Since 2017 Vice President, Science & Education, The Field Museum, Chicago. USA 2014-2017 Director, Integrative Research Center, Science & Education, The Field Museum, Chicago, USA. Since 2014 Curator, Integrative Research Center, Science & Education, The Field Museum, Chicago, USA. 2013-2014 Associate Director, Integrative Research Center, Science & Education, The Field Museum, Chicago, USA. 2009-2013 Chair, Dept. of Botany, The Field Museum, Chicago, USA. Since 2011 MacArthur Associate Curator, Dept. of Botany, The Field Museum, Chicago, USA. 2006-2014 Associate Curator, Dept. of Botany, The Field Museum, Chicago, USA. 2005-2009 Head of Cryptogams, Dept. of Botany, The Field Museum, Chicago, USA. Since 2004 Member, Committee on Evolutionary Biology, University of Chicago. Courses: BIOS 430 Evolution (UIC), BIOS 23410 Complex Interactions: Coevolution, Parasites, Mutualists, and Cheaters (U of C) Reading group: Phylogenetic methods. 2003-2006 Assistant Curator, Dept. of Botany, The Field Museum, Chicago, USA. 1998-2003 Privatdozent (Assistant Professor), Botanical Institute, University – GHS - Essen. Lectures: General Botany, Evolution of lower plants, Photosynthesis, Courses: Cryptogams, Biology -

<I> Myriospora</I> (<I>Acarosporaceae</I>)
MYCOTAXON ISSN (print) 0093-4666 (online) 2154-8889 Mycotaxon, Ltd. ©2017 October–December 2017—Volume 132, pp. 857–865 https://doi.org/10.5248/132.857 New reports of Myriospora (Acarosporaceae) from Europe Kerry Knudsen1, Jana Kocourková1 & Ulf Schiefelbein2 1 Czech University of Life Sciences Prague, Faculty of Environmental Sciences, Department of Ecology, Kamýcká 129, Praha 6 - Suchdol, CZ–165 21, Czech Republic 2 Blücherstraße 71, D-18055 Rostock, Germany * Correspondence to: [email protected] Abstract—Myriospora dilatata is newly reported for the Czech Republic and M. myochroa new for Italy. Myriospora rufescens was rediscovered in Germany almost 100 years after its first collection. A neotype is designated for Acarospora fusca, which is recognized as a synonym of M. rufescens. Key words—Myriospora hassei, Silobia, Trimmatothelopsis Introduction The genus Myriospora in the Acarosporaceae is a well-supported clade distinguished by a constellation of morphological characters (non-lecideine apothecia, high hymenium, thin paraphyses, interrupted algal layer, short conidia, no secondary metabolites or norstictic acid) (Wedin et al. 2009; Westberg et al. 2011, 2015). The genus currently contains 12 species that occur in Antarctica, Asia, Europe, and North and South America (Knudsen 2011, Westberg et al. 2011, Knudsen et al. 2012, Knudsen & Bungartz 2014, Schiefelbein et al. 2015, Purvis et al. in press). Myriospora fulvoviridula (Harm.) Cl. Roux is a synonym of M. scabrida (H. Magn.) K. Knudsen & Arcadia (Knudsen et al. 2017, Roux et al. 2014). The most common species in the genus is M. smaragdula (Wahlenb.) Nägeli ex Uloth, which occurs in Asia, Europe, North and South America (Magnusson 1929, Knudsen 2007, Westberg et al.