Mitogenic Stimulation Accelerates Influenza-Induced Mortality By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Postviral Bronchial Hyperreactivity Syndrome: Recognizing Asthma's
• Postviral bronchial hyperreactivity syndrome: Recognizing asthmas great mimic DAVID OSTRANSKY, DO FRANCIS X. BLAIS, DO Although there are no prospec- (Key words: Viral infections, asthma, tive studies regarding the frequency of respiratory tract infections, postviral postviral bronchial hyperreactivity syn- bronchial hyperreactivity syndrome) drome, it is a common complication of upper and lower respiratory tract viral Viral respiratory tract infections frequently infections. The respiratory symptoms cause wheezing and other asthmalike symp- closely resemble those of asthma, but they toms. Several investigators have demonstrated are present for only 3 weeks to 3 months pertinent features of this abbreviated form of following the acute infection phase. Defin- asthma, including early response phase, late ing the mechanisms of this syndrome may response phase, and bronchial hyperreac- provide insight into the pathogenesis of tivity. 1-5 Understanding the mechanisms by asthma. Postviral bronchial hyperreac- which viral respiratory tract infections precipi- tivity syndrome is frequently misdiag- tate the airway abnormalities of asthma may nosed and inappropriately managed be- be a potential key to the pathogenesis of cause many physicians are unfamiliar asthma, although whether viral respiratory with this illness. Because of its character- tract infections truly cause asthma is un- istic history, diagnosis is straightforward proved.6 when the physician knows what to look The symptom complex following viral up- for, and response to therapy is excellent. per or lower respiratory tract infections (or This report presents a case history fol- both) has not been formally identified. It is fre- lowed by a review of the proposed mecha- quently misdiagnosed by physicians who then nisms of bronchial hyperreactivity follow- institute inappropriate diagnostic studies and ing viral respiratory infections. -

COVID-19 Pneumonia: the Great Radiological Mimicker
Duzgun et al. Insights Imaging (2020) 11:118 https://doi.org/10.1186/s13244-020-00933-z Insights into Imaging EDUCATIONAL REVIEW Open Access COVID-19 pneumonia: the great radiological mimicker Selin Ardali Duzgun* , Gamze Durhan, Figen Basaran Demirkazik, Meltem Gulsun Akpinar and Orhan Macit Ariyurek Abstract Coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has rapidly spread worldwide since December 2019. Although the reference diagnostic test is a real-time reverse transcription-polymerase chain reaction (RT-PCR), chest-computed tomography (CT) has been frequently used in diagnosis because of the low sensitivity rates of RT-PCR. CT fndings of COVID-19 are well described in the literature and include predominantly peripheral, bilateral ground-glass opacities (GGOs), combination of GGOs with consolida- tions, and/or septal thickening creating a “crazy-paving” pattern. Longitudinal changes of typical CT fndings and less reported fndings (air bronchograms, CT halo sign, and reverse halo sign) may mimic a wide range of lung patholo- gies radiologically. Moreover, accompanying and underlying lung abnormalities may interfere with the CT fndings of COVID-19 pneumonia. The diseases that COVID-19 pneumonia may mimic can be broadly classifed as infectious or non-infectious diseases (pulmonary edema, hemorrhage, neoplasms, organizing pneumonia, pulmonary alveolar proteinosis, sarcoidosis, pulmonary infarction, interstitial lung diseases, and aspiration pneumonia). We summarize the imaging fndings of COVID-19 and the aforementioned lung pathologies that COVID-19 pneumonia may mimic. We also discuss the features that may aid in the diferential diagnosis, as the disease continues to spread and will be one of our main diferential diagnoses some time more. -

Pediatric Ambulatory Community Acquired Pneumonia (CAP)
ANMC Pediatric (≥3mo) Ambulatory Community Acquired Pneumonia (CAP) Treatment Guideline Criteria for Respiratory Distress Criteria For Outpatient Management Testing/Imaging for Outpatient Management Tachypnea, in breaths/min: Mild CAP: no signs of respiratory distress Vital Signs: Standard VS and Pulse Oximetry Age 0-2mo: >60 Able to tolerate PO Labs: No routine labs indicated Age 2-12mo: >50 No concerns for pathogen with increased virulence Influenza PCR during influenza season Age 1-5yo: >40 (ex. CA-MRSA) Blood cultures if not fully immunized OR fails to Age >5yo: >20 Family able to carefully observe child at home, comply improve/worsens after initiation of antibiotics Dyspnea with therapy plan, and attend follow up appointments Urinary antigen detection testing is not Retractions recommended in children; false-positive tests are common. Grunting If patient does not meet outpatient management criteria Radiography: No routine CXR indicated Nasal flaring refer to inpatient pneumonia guideline for initial workup Apnea and testing. AP and lateral CXR if fails initial antibiotic therapy Altered mental status AP and lateral CXR 4-6 weeks after diagnosis if Pulse oximetry <90% on room air recurrent pneumonia involving the same lobe Treatment Selection Suspected Viral Pneumonia Most Common Pathogens: Influenza A & B, Adenovirus, Respiratory Syncytial Virus, Parainfluenza No antimicrobial therapy is necessary. Most common in <5yo If influenza positive, see influenza guidelines for treatment algorithm. Suspected Bacterial -

Community-Acquired Pneumonia in Children KIMBERLY STUCKEY-SCHROCK, MD, Memphis, Tennessee BURTON L
Community-Acquired Pneumonia in Children KIMBERLY STUCKEY-SCHROCK, MD, Memphis, Tennessee BURTON L. HAYES, MD, and CHRISTA M. GEORGE, PharmD University of Tennessee Health Science Center, Memphis, Tennessee Community-acquired pneumonia is a potentially serious infection in children and often results in hospitalization. The diagnosis can be based on the history and physical examination results in children with fever plus respiratory signs and symptoms. Chest radiography and rapid viral testing may be helpful when the diagnosis is unclear. The most likely etiology depends on the age of the child. Viral and Streptococcus pneumoniae infections are most common in preschool-aged children, whereas Mycoplasma pneumoniae is common in older children. The decision to treat with antibiotics is challenging, especially with the increasing prevalence of viral and bacterial coinfections. Preschool-aged children with uncomplicated bacterial pneumonia should be treated with amoxicillin. Macrolides are first-line agents in older children. Immunization with the 13-valent pneumococcal conjugate vaccine is important in reducing the severity of childhood pneumococcal infections. (Am Fam Physician. 2012;86(7):661-667. Copyright © 2012 American Academy of Family Physicians.) ommunity-acquired pneumonia infection accounts for 30 to 50 percent of CAP (CAP) is a significant cause of infections in children.7 respiratory morbidity and mor- Streptococcus pneumoniae is the most com- tality in children, especially in mon bacterial cause of CAP. The widespread C developing countries.1 Worldwide, CAP is the use of pneumococcal immunization has leading cause of death in children younger reduced the incidence of invasive disease.8 than five years.2 Factors that increase the Children with underlying conditions and incidence and severity of pneumonia in chil- those who attend child care are at higher risk dren include prematurity, malnutrition, low of invasive pneumococcal disease. -

Aspergillosis Complicating Severe Coronavirus Disease Kieren A
SYNOPSIS Aspergillosis Complicating Severe Coronavirus Disease Kieren A. Marr, Andrew Platt, Jeffrey A. Tornheim, Sean X. Zhang, Kausik Datta, Celia Cardozo, Carolina Garcia-Vidal describing epidemiology and significance of aspergil- Aspergillosis complicating severe influenza infection losis occurring after severe viral infections, especially has been increasingly detected worldwide. Recently, coronavirus disease–associated pulmonary aspergil- influenza and coronavirus disease (COVID-19). losis (CAPA) has been detected through rapid reports, Aspergillosis associated with severe influenza primarily from centers in Europe. We provide a case virus infection (influenza-associated aspergillosis, series of CAPA, adding 20 cases to the literature, with IAA) was reported in 1951, when Abbott et al. de- review of pathophysiology, diagnosis, and outcomes. scribed fatal infection in a woman with cavitary in- The syndromes of pulmonary aspergillosis complicating vasive pulmonary aspergillosis noted on autopsy (2). severe viral infections are distinct from classic invasive Scattered reports appeared in thereafter; Adalja et al. aspergillosis, which is recognized most frequently in summarized 27 cases in the literature during 1952– persons with neutropenia and in other immunocompro- 2011, which reported predominance after influenza mised persons. Combined with severe viral infection, A infection, associated lymphopenia, and occurring aspergillosis comprises a constellation of airway-inva- in persons of a broad age range (14–89 years), but sive and angio-invasive disease and results in risks as- sociated with poor airway fungus clearance and killing, with little underlying lung disease (3). There were including virus- or inflammation-associated epithelial increased numbers of cases reported during and af- damage, systemic immunosuppression, and underlying ter the 2009 influenza A(H1N1) pandemic (3–10). -

IDSA/ATS Consensus Guidelines on The
SUPPLEMENT ARTICLE Infectious Diseases Society of America/American Thoracic Society Consensus Guidelines on the Management of Community-Acquired Pneumonia in Adults Lionel A. Mandell,1,a Richard G. Wunderink,2,a Antonio Anzueto,3,4 John G. Bartlett,7 G. Douglas Campbell,8 Nathan C. Dean,9,10 Scott F. Dowell,11 Thomas M. File, Jr.12,13 Daniel M. Musher,5,6 Michael S. Niederman,14,15 Antonio Torres,16 and Cynthia G. Whitney11 1McMaster University Medical School, Hamilton, Ontario, Canada; 2Northwestern University Feinberg School of Medicine, Chicago, Illinois; 3University of Texas Health Science Center and 4South Texas Veterans Health Care System, San Antonio, and 5Michael E. DeBakey Veterans Affairs Medical Center and 6Baylor College of Medicine, Houston, Texas; 7Johns Hopkins University School of Medicine, Baltimore, Maryland; 8Division of Pulmonary, Critical Care, and Sleep Medicine, University of Mississippi School of Medicine, Jackson; 9Division of Pulmonary and Critical Care Medicine, LDS Hospital, and 10University of Utah, Salt Lake City, Utah; 11Centers for Disease Control and Prevention, Atlanta, Georgia; 12Northeastern Ohio Universities College of Medicine, Rootstown, and 13Summa Health System, Akron, Ohio; 14State University of New York at Stony Brook, Stony Brook, and 15Department of Medicine, Winthrop University Hospital, Mineola, New York; and 16Cap de Servei de Pneumologia i Alle`rgia Respirato`ria, Institut Clı´nic del To`rax, Hospital Clı´nic de Barcelona, Facultat de Medicina, Universitat de Barcelona, Institut d’Investigacions Biome`diques August Pi i Sunyer, CIBER CB06/06/0028, Barcelona, Spain. EXECUTIVE SUMMARY priate starting point for consultation by specialists. Substantial overlap exists among the patients whom Improving the care of adult patients with community- these guidelines address and those discussed in the re- acquired pneumonia (CAP) has been the focus of many cently published guidelines for health care–associated different organizations, and several have developed pneumonia (HCAP). -

Radiologically Suspected Organizing Pneumonia in a Patient Recovering from COVID-19: a Case Report
Infect Chemother. 2021 Mar;53(1):e8 https://doi.org/10.3947/ic.2021.0013 pISSN 2093-2340·eISSN 2092-6448 Case Report Radiologically Suspected Organizing Pneumonia in a Patient Recovering from COVID-19: A Case Report Hyeonji Seo 1, Jiwon Jung 1, Min Jae Kim 1, Se Jin Jang 2, and Sung-Han Kim 1 1Department of Infectious Diseases, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea 2Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea Received: Jan 28, 2021 ABSTRACT Accepted: Feb 10, 2021 Corresponding Author: We report a case of coronavirus disease 2019 (COVID-19)-associated radiologically suspected Sung-Han Kim, MD organizing pneumonia with repeated negative Severe acute respiratory syndrome coronavirus Department of Infectious Diseases, Asan 2 (SARS-CoV-2) polymerase chain reaction (PCR) results from nasopharyngeal swab Medical Center, University of Ulsan College of and sputum samples, but positive result from bronchoalveolar lavage fluid. Performing Medicine, 88, Olympic-ro, 43-gil, Songpa-gu, SARS-CoV-2 RT-PCR in upper respiratory tract samples only could fail to detect COVID-19- Seoul 05505, Korea. Tel: +82-2-3010-3305 associated pneumonia, and SARS-CoV-2 could be an etiology of radiologically suspected Fax: +82-2-3010-6970 organizing pneumonia. E-mail: [email protected] Keywords: COVID-19; SARS-CoV-2; Organizing pneumonia; Bronchoalveolar lavage; Copyright © 2021 by The Korean Society Polymerase chain reaction of Infectious Diseases, Korean Society for Antimicrobial -

Introduction to Respiratory Diseases
Case Studies: Introduction to Respiratory Diseases Respiratory tract infections are a major reason why children and the elderly seek medical care. These infections are more common in cold-weather months in locales with temperate climates. Respiratory tract infections are primarily spread by inhalation of aerosolized respiratory secretions from infected hosts. Some respiratory tract pathogens such as rhinoviruses can also be spread by direct contact with mucous membranes, but this mode of transmission is much less common than inhalation. For the purpose of our discussion, we will divide these types of infection into two groups, upper tract and lower tract infection. The most common form of upper respiratory tract infection is pharyngitis. Pharyngitis is seen most frequently in children from 2 years of age through adolescence. The most common etiologic agents of pharyngitis are viruses, particularly adenoviruses, and group A streptococci. Pharyngitis due to group A streptococci predisposes individuals to the development of the poststreptococcal sequela rheumatic fever. Because this sequela can be prevented by penicillin treatment, aggressive diagnosis and treatment of group A streptococcal pharyngitis is needed. Otitis media is a common infectious problem in infants and young children. The most frequently encountered agents of this infection are bacterial, with Streptococcus pneumoniae, non-typeable Haemophilus influenzae, and Moraxella catarrhalis being most common. These organisms, along with certain viruses and anaerobic bacteria from the oral cavity, are the most important pathogens in sinusitis. S. pneumoniae, H. influenzae, Moraxella catarrhalis, and adenoviruses, as well as Chlamydia trachomtis in neonates, are the common etiologic agents of conjunctivitis. External otitis, a common problem in swimmers, is more common in warm weather months. -

Supplement 1 READ Code Description Disease Group Disease Further Specified 1652 Feels Hot/Feverish OTHER OTHER 1653 Fever with S
Supplement 1 Disease further READ code Description Disease group specified 1652 Feels hot/feverish OTHER OTHER 1653 Fever with sweating OTHER OTHER 1712 Dry cough LRTI LRTI - unspecified 1713 Productive cough -clear sputum LRTI LRTI - unspecified 1714 Productive cough -green sputum LRTI LRTI - unspecified 1715 Productive cough-yellow sputum LRTI LRTI - unspecified 1716 Productive cough NOS LRTI LRTI - unspecified 1716.11 Coughing up phlegm LRTI LRTI - unspecified 1717 Night cough present LRTI LRTI - unspecified 1719 Chesty cough LRTI LRTI - unspecified 1719.11 Bronchial cough LRTI LRTI - unspecified 165..11 Fever symptoms OTHER OTHER 165..12 Pyrexia symptoms OTHER OTHER 16L..00 Influenza-like symptoms LRTI INFLUENZA 17...00 Respiratory symptoms LRTI LRTI - unspecified 171..00 Cough LRTI LRTI - unspecified 171..11 C/O - cough LRTI LRTI - unspecified 171A.00 Chronic cough LRTI LRTI - unspecified 171B.00 Persistent cough LRTI LRTI - unspecified 171C.00 Morning cough LRTI LRTI - unspecified 171D.00 Evening cough LRTI LRTI - unspecified 171E.00 Unexplained cough LRTI LRTI - unspecified 171F.00 Cough with fever LRTI LRTI - unspecified 171G.00 Bovine cough LRTI LRTI - unspecified 171H.00 Difficulty in coughing up sputum LRTI LRTI - unspecified 171J.00 Reflux cough LRTI LRTI - unspecified 171K.00 Barking cough LRTI LRTI - unspecified 171L.00 Cough on exercise LRTI LRTI - unspecified 171Z.00 Cough symptom NOS LRTI LRTI - unspecified 173A.00 Exercise induced asthma ASTHMA ASTHMA 173c.00 Occupational asthma ASTHMA ASTHMA 173d.00 Work aggravated asthma -
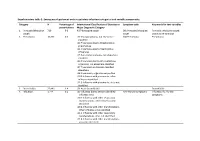
Supplementary Table 1. Emergency Department Acute Respiratory Infections Categories and Variable Components
Supplementary table 1. Emergency department acute respiratory infections categories and variable components Category N Percentage of International Classification of Diseases or Symptom code Key words for text variables presentations Major Diagnostic Category 1. Pertussis/Whooping 710 0.0 A37 Whooping cough SNJ Pertussis/whooping Pertussis; whooping cough; cough cough post-tussive vomiting 2. Pneumonia 10,322 0.6 J12 Viral pneumonia, not elsewhere SQJ Pneumonia Pneumonia classified J13 Pneumonia due to Streptococcus pneumoniae J14 Pneumonia due to Haemophilus influenzae J15 Bacterial pneumonia, not elsewhere classified J16 Pneumonia due to other infectious organisms, not elsewhere classified J17 Pneumonia in diseases classified elsewhere J18 Pneumonia, organism unspecified J10.0 Influenza with pneumonia, other influenza identified J11.0 Influenza with pneumonia, virus not identified 3. Bronchiolitis 22,446 1.4 J21 Acute bronchiolitis Bronchiolitis 4. Influenza 1,774 0.1 J09 Influenza due to certain identified AAV Flu Like Symptoms Influenza; flu; flu-like influenza virus symptoms J10.1 Influenza with other respiratory manifestations, other influenza virus identified J10.8 Influenza with other manifestations, other influenza virus identified J11.1 Influenza with other respiratory manifestations, virus not identified J11.8 Influenza with other manifestations, virus not identified Category N Percentage of International Classification of Diseases or Symptom code Key words for text variables presentations Major Diagnostic Category 5. Unspecified ALRI 3,780 0.2 J22 Unspecified acute lower respiratory SQD Chest infection Unspecified Acute Lower (includes chest infection infection Respiratory Infection; LRTI; & LRTI) lower respiratory tract infection; chest infection 6. Bronchitis 1,234 0.1 J20 Acute bronchitis SQC Bronchitis Bronchitis J40 Bronchitis, not specified as acute or chronic 7. -

An Uncommon Case of Secondary Organizing Pneumonia Due to Influenza Type B
Case Report An Uncommon Case of Secondary Organizing Pneumonia Due to Influenza Type B Parth Shah 1, Philip T. Sobash 1,* , Krishna Vedala 1 , Krishna Kakkera 2 and Gilbert-Roy Kamoga 1 1 Internal Medicine, White River Health System, Batesville, AR 72501, USA; [email protected] (P.S.); [email protected] (K.V.); [email protected] (G.-R.K.) 2 Pulmonology and Critical Care, White River Health System, Batesville, AR 72501, USA; [email protected] * Correspondence: [email protected] Received: 17 December 2020; Accepted: 22 February 2021; Published: 10 March 2021 Abstract: Secondary organizing pneumonia refers to a disease process caused by pulmonary tissue injury. Various insults can cause secondary organizing pneumonia, including multiple types of infections and cancer. The mainstay of diagnosis is a combination of imaging and lung biopsy showing inflammatory changes, specifically plugs with granulated tissue and fibrosis. Clinical suspicion needs to be raised for secondary organizing pneumonia when a patient is requiring increasing amounts of oxygen in the presence of treatment for pneumonia or another underlying lung disease. Here, we present the case of a 65-year-old male who presented with acute hypoxemic respiratory failure in the setting of previously having been tested positive for influenza B. Aggressive steroids with eventual tapering of his O2 requirements led to a successful outcome. While influenza has been reported as a cause of secondary organizing pneumonia after proceeding infection, these cases are usually represented by type A, rather than B. Keywords: secondary organizing pneumonia; influenza B; viral pneumonia 1. Introduction Organizing pneumonia (OP) is a clinicopathological syndrome that is comprised of systemic symptoms with consolidations on imaging with restrictive deficits on PFTs and granulation tissues found in the airways [1]. -

Lung Infections C 4 Pathogenesis of Lower Respiratory Tract
302 Thorax 1998;53:302–307 Lung infections c 4 Series editor: S L Hill Thorax: first published as 10.1136/thx.53.4.302 on 1 April 1998. Downloaded from Pathogenesis of lower respiratory tract infections due to Chlamydia, Mycoplasma, Legionella and viruses Paul Andersen Acute infection of the lower respiratory tract terminal organelle.18 Metabolic and ultrastruc- comprises bronchitis, bronchiolitis, and pneu- tural alterations in the aVected cell are seen and monia. From a clinical point of view it may be these result in epithelial cell damage and cilio- diYcult to distinguish these disease entities stasis. Some epithelial cell lines produce and one infection may progress into another. cytokines when stimulated with other bacteria The most common pathogens causing these such as Escherichia coli,21 and epithelial cells infections are the primary respiratory viruses might therefore play a more active role in the (respiratory syncytial virus (RSV), influenza mucosal immune response after extracellular virus, adenoviruses, parainfluenza virus, and bacterial infection. In acute respiratory viral rhinovirus12), Mycoplasma pneumoniae,3 and diseases a number of diVerent inflammatory Chlamydia species.4–6 Legionella may cause mediators such as kinins and cytokines have pneumonia and non-pneumonic upper respira- also been demonstrated. In some infections tory tract infection and approximately 85% of c such as influenza extensive infiltrations with ases are caused by L pneumophila. Long lasting polymorphonuclear leucocytes (PMN), sequelae such as bronchiectasis,