Distinct Transcriptional Programming Drive Response to MAPK Inhibition in BRAF -Mutant Melanoma Patient-Derived Xenografts
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Regulatory Roles of Phosphatases in Cancer
Oncogene (2014) 33, 939–953 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW The regulatory roles of phosphatases in cancer J Stebbing1, LC Lit1, H Zhang, RS Darrington, O Melaiu, B Rudraraju and G Giamas The relevance of potentially reversible post-translational modifications required for controlling cellular processes in cancer is one of the most thriving arenas of cellular and molecular biology. Any alteration in the balanced equilibrium between kinases and phosphatases may result in development and progression of various diseases, including different types of cancer, though phosphatases are relatively under-studied. Loss of phosphatases such as PTEN (phosphatase and tensin homologue deleted on chromosome 10), a known tumour suppressor, across tumour types lends credence to the development of phosphatidylinositol 3--kinase inhibitors alongside the use of phosphatase expression as a biomarker, though phase 3 trial data are lacking. In this review, we give an updated report on phosphatase dysregulation linked to organ-specific malignancies. Oncogene (2014) 33, 939–953; doi:10.1038/onc.2013.80; published online 18 March 2013 Keywords: cancer; phosphatases; solid tumours GASTROINTESTINAL MALIGNANCIES abs in sera were significantly associated with poor survival in Oesophageal cancer advanced ESCC, suggesting that they may have a clinical utility in Loss of PTEN (phosphatase and tensin homologue deleted on ESCC screening and diagnosis.5 chromosome 10) expression in oesophageal cancer is frequent, Cao et al.6 investigated the role of protein tyrosine phosphatase, among other gene alterations characterizing this disease. Zhou non-receptor type 12 (PTPN12) in ESCC and showed that PTPN12 et al.1 found that overexpression of PTEN suppresses growth and protein expression is higher in normal para-cancerous tissues than induces apoptosis in oesophageal cancer cell lines, through in 20 ESCC tissues. -

Curcumin Alters Gene Expression-Associated DNA Damage, Cell Cycle, Cell Survival and Cell Migration and Invasion in NCI-H460 Human Lung Cancer Cells in Vitro
ONCOLOGY REPORTS 34: 1853-1874, 2015 Curcumin alters gene expression-associated DNA damage, cell cycle, cell survival and cell migration and invasion in NCI-H460 human lung cancer cells in vitro I-TSANG CHIANG1,2, WEI-SHU WANG3, HSIN-CHUNG LIU4, SU-TSO YANG5, NOU-YING TANG6 and JING-GUNG CHUNG4,7 1Department of Radiation Oncology, National Yang‑Ming University Hospital, Yilan 260; 2Department of Radiological Technology, Central Taiwan University of Science and Technology, Taichung 40601; 3Department of Internal Medicine, National Yang‑Ming University Hospital, Yilan 260; 4Department of Biological Science and Technology, China Medical University, Taichung 404; 5Department of Radiology, China Medical University Hospital, Taichung 404; 6Graduate Institute of Chinese Medicine, China Medical University, Taichung 404; 7Department of Biotechnology, Asia University, Taichung 404, Taiwan, R.O.C. Received March 31, 2015; Accepted June 26, 2015 DOI: 10.3892/or.2015.4159 Abstract. Lung cancer is the most common cause of cancer CARD6, ID1 and ID2 genes, associated with cell survival and mortality and new cases are on the increase worldwide. the BRMS1L, associated with cell migration and invasion. However, the treatment of lung cancer remains unsatisfactory. Additionally, 59 downregulated genes exhibited a >4-fold Curcumin has been shown to induce cell death in many human change, including the DDIT3 gene, associated with DNA cancer cells, including human lung cancer cells. However, the damage; while 97 genes had a >3- to 4-fold change including the effects of curcumin on genetic mechanisms associated with DDIT4 gene, associated with DNA damage; the CCPG1 gene, these actions remain unclear. Curcumin (2 µM) was added associated with cell cycle and 321 genes with a >2- to 3-fold to NCI-H460 human lung cancer cells and the cells were including the GADD45A and CGREF1 genes, associated with incubated for 24 h. -

Targeting Protein Tyrosine Phosphatases in Cancer Lakshmi Reddy Bollu, Abhijit Mazumdar, Michelle I
Published OnlineFirst January 13, 2017; DOI: 10.1158/1078-0432.CCR-16-0934 Molecular Pathways Clinical Cancer Research Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer Lakshmi Reddy Bollu, Abhijit Mazumdar, Michelle I. Savage, and Powel H. Brown Abstract The aberrant activation of oncogenic signaling pathways is a act as tumor suppressor genes by terminating signal responses universal phenomenon in cancer and drives tumorigenesis and through the dephosphorylation of oncogenic kinases. More malignant transformation. This abnormal activation of signal- recently, it has become clear that several PTPs overexpressed ing pathways in cancer is due to the altered expression of in human cancers do not suppress tumor growth; instead, they protein kinases and phosphatases. In response to extracellular positively regulate signaling pathways and promote tumor signals, protein kinases activate downstream signaling path- development and progression. In this review, we discuss both ways through a series of protein phosphorylation events, ulti- types of PTPs: those that have tumor suppressor activities as mately producing a signal response. Protein tyrosine phospha- well as those that act as oncogenes. We also discuss the tases (PTP) are a family of enzymes that hydrolytically remove potential of PTP inhibitors for cancer therapy. Clin Cancer Res; phosphate groups from proteins. Initially, PTPs were shown to 23(9); 1–7. Ó2017 AACR. Background in cancer and discuss the current status of PTP inhibitors for cancer therapy. Signal transduction is a complex process that transmits extra- PTPs belong to a superfamily of enzymes that hydrolytically cellular signals effectively through a cascade of events involving remove phosphate groups from proteins (2). -

Bortezomib Inhibits Lung Fibrosis and Fibroblast Activation Without Proteasome
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.26.433086; this version posted February 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Bortezomib inhibits lung fibrosis and fibroblast activation without proteasome 2 inhibition 3 4 Loka Raghu Kumar Penke, Jennifer Speth, Scott Wettlaufer, Christina Draijer and Marc 5 Peters-Golden1* 6 7 Affiliations 8 Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine, 9 University of Michigan, Ann Arbor, Michigan, USA. 10 1 Graduate Program in Immunology, University of Michigan, Ann Arbor, Michigan, USA. 11 12 Correspondence: 13 Marc Peters-Golden, M.D. 14 6301 MSRBIII, 1150 W. Medical Center Drive, Ann Arbor, MI 48109-5642 15 Tel: 734-936-5047 16 Fax: 734-764-4556 17 Email: [email protected] 18 Author Contributions: L.R.K.P. planned and performed experiments, analyzed the 19 data, organized data for presentation, and wrote the manuscript. J.M.S, S.H.W and C.D 20 performed experiments. M.P.-G. planned experiments, analyzed data, and wrote the 21 manuscript. 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.02.26.433086; this version posted February 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

GATA2 Regulates Mast Cell Identity and Responsiveness to Antigenic Stimulation by Promoting Chromatin Remodeling at Super- Enhancers
ARTICLE https://doi.org/10.1038/s41467-020-20766-0 OPEN GATA2 regulates mast cell identity and responsiveness to antigenic stimulation by promoting chromatin remodeling at super- enhancers Yapeng Li1, Junfeng Gao 1, Mohammad Kamran1, Laura Harmacek2, Thomas Danhorn 2, Sonia M. Leach1,2, ✉ Brian P. O’Connor2, James R. Hagman 1,3 & Hua Huang 1,3 1234567890():,; Mast cells are critical effectors of allergic inflammation and protection against parasitic infections. We previously demonstrated that transcription factors GATA2 and MITF are the mast cell lineage-determining factors. However, it is unclear whether these lineage- determining factors regulate chromatin accessibility at mast cell enhancer regions. In this study, we demonstrate that GATA2 promotes chromatin accessibility at the super-enhancers of mast cell identity genes and primes both typical and super-enhancers at genes that respond to antigenic stimulation. We find that the number and densities of GATA2- but not MITF-bound sites at the super-enhancers are several folds higher than that at the typical enhancers. Our studies reveal that GATA2 promotes robust gene transcription to maintain mast cell identity and respond to antigenic stimulation by binding to super-enhancer regions with dense GATA2 binding sites available at key mast cell genes. 1 Department of Immunology and Genomic Medicine, National Jewish Health, Denver, CO 80206, USA. 2 Center for Genes, Environment and Health, National Jewish Health, Denver, CO 80206, USA. 3 Department of Immunology and Microbiology, University of Colorado Anschutz Medical Campus, Aurora, ✉ CO 80045, USA. email: [email protected] NATURE COMMUNICATIONS | (2021) 12:494 | https://doi.org/10.1038/s41467-020-20766-0 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-20766-0 ast cells (MCs) are critical effectors in immunity that at key MC genes. -

Dual-Specificity Phosphatases in Immunity and Infection
International Journal of Molecular Sciences Review Dual-Specificity Phosphatases in Immunity and Infection: An Update Roland Lang * and Faizal A.M. Raffi Institute of Clinical Microbiology, Immunology and Hygiene, Universitätsklinikum Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91054 Erlangen, Germany * Correspondence: [email protected]; Tel.: +49-9131-85-22979 Received: 15 May 2019; Accepted: 30 May 2019; Published: 2 June 2019 Abstract: Kinase activation and phosphorylation cascades are key to initiate immune cell activation in response to recognition of antigen and sensing of microbial danger. However, for balanced and controlled immune responses, the intensity and duration of phospho-signaling has to be regulated. The dual-specificity phosphatase (DUSP) gene family has many members that are differentially expressed in resting and activated immune cells. Here, we review the progress made in the field of DUSP gene function in regulation of the immune system during the last decade. Studies in knockout mice have confirmed the essential functions of several DUSP-MAPK phosphatases (DUSP-MKP) in controlling inflammatory and anti-microbial immune responses and support the concept that individual DUSP-MKP shape and determine the outcome of innate immune responses due to context-dependent expression and selective inhibition of different mitogen-activated protein kinases (MAPK). In addition to the canonical DUSP-MKP, several small-size atypical DUSP proteins regulate immune cells and are therefore also reviewed here. Unexpected and complex findings in DUSP knockout mice pose new questions regarding cell type-specific and redundant functions. Another emerging question concerns the interaction of DUSP-MKP with non-MAPK binding partners and substrate proteins. -

Dual-Specificity Phosphatases 2
Genes and Immunity (2013) 14, 1–6 & 2013 Macmillan Publishers Limited All rights reserved 1466-4879/13 www.nature.com/gene REVIEW Dual-specificity phosphatases 2: surprising positive effect at the molecular level and a potential biomarker of diseases WWei1,2, Y Jiao2,3, A Postlethwaite4,5, JM Stuart4,5, Y Wang6, D Sun1 and W Gu2 Dual-specificity phosphatases (DUSPs) is an emerging subclass of the protein tyrosine phosphatase gene superfamily, a heterogeneous group of protein phosphatases that can dephosphorylate both phosphotyrosine and phosphoserine/ phosphothreonine residues within the one substrate. Recently, a series of investigations of DUSPs defined their essential roles in cell proliferation, cancer and the immune response. This review will focus on DUSP2, its involvement in different diseases and its potential as a therapeutic target. Genes and Immunity (2013) 14, 1–6; doi:10.1038/gene.2012.54; published online 29 November 2012 Keywords: dual-specificity phosphatases; disease; mitogen-activated protein kinase; immune INTRODUCTION extracellular stimuli. Inducible nucleuses MKPs include DUSP1, Mitogen-activated protein kinase (MAPK) activation cascades DUSP2, DUSP4 and DUSP5, which originated from a common mediate various physiological processes, such as cell proliferation, ancestral gene. They were shown to dephosphorylate Erks, Jnk differentiation, stress responses, inflammation, apoptosis and and p38 MAPKs to the same extent and to be induced by growth immune defense.1–4 Dysregulation of MAPK activation cascades or stress signals. DUSP6, DUSP7 and DUSP9 are cytoplasmic Erk- has been implicated in various diseases and has been the focus of specific MPKs, which can preferentially recognize Erk1 and Erk2 extensive research.5–7 MAPKs are grouped into three major classes in vitro. -

HER2 Therapy in Breast Cancer
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 44), pp: 77207-77218 Research Paper DUSP4 is associated with increased resistance against anti- HER2 therapy in breast cancer Otília Menyhart1, Jan Budczies2, Gyöngyi Munkácsy1, Francisco J. Esteva3, András Szabó1, Teresa Puig Miquel4 and Balázs Győrffy1,5 1Semmelweis University 2nd Department of Pediatrics, Budapest, Hungary 2Institute of Pathology, Charité University Hospital, Berlin, Germany 3Clinical Cancer Center, NYU Langone Medical Center, New York, NY, USA 4New Terapeutics Targets Laboratory (TargetsLab), Department of Medical Sciences, University of Girona, Girona, Spain 5MTA TTK Lendület Cancer Biomarker Research Group, Institute of Enzymology, Budapest, Hungary Correspondence to: Balázs Győrffy, email: [email protected] Keywords: DUSP4, targeted therapy, trastuzumab, biomarker, breast cancer Received: March 09, 2017 Accepted: June 27, 2017 Published: August 24, 2017 Copyright: Menyhart et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT The majority of patients develop resistance against suppression of HER2-signaling mediated by trastuzumab in HER2 positive breast cancer (BC). HER2 overexpression activates multiple signaling pathways, including the mitogen-activated protein kinase (MAPK) cascade. MAPK phosphatases (MKPs) are essential regulators of MAPKs and participate in many facets of cellular regulation, including proliferation and apoptosis. We aimed to identify whether differential MKPs are associated with resistance to targeted therapy in patients previously treated with trastuzumab. Using gene chip data of 88 HER2-positive, trastuzumab treated BC patients, candidate MKPs were identified by Receiver Operator Characteristics analysis performed in R. -

Live-Cell Imaging Rnai Screen Identifies PP2A–B55α and Importin-Β1 As Key Mitotic Exit Regulators in Human Cells
LETTERS Live-cell imaging RNAi screen identifies PP2A–B55α and importin-β1 as key mitotic exit regulators in human cells Michael H. A. Schmitz1,2,3, Michael Held1,2, Veerle Janssens4, James R. A. Hutchins5, Otto Hudecz6, Elitsa Ivanova4, Jozef Goris4, Laura Trinkle-Mulcahy7, Angus I. Lamond8, Ina Poser9, Anthony A. Hyman9, Karl Mechtler5,6, Jan-Michael Peters5 and Daniel W. Gerlich1,2,10 When vertebrate cells exit mitosis various cellular structures can contribute to Cdk1 substrate dephosphorylation during vertebrate are re-organized to build functional interphase cells1. This mitotic exit, whereas Ca2+-triggered mitotic exit in cytostatic-factor- depends on Cdk1 (cyclin dependent kinase 1) inactivation arrested egg extracts depends on calcineurin12,13. Early genetic studies in and subsequent dephosphorylation of its substrates2–4. Drosophila melanogaster 14,15 and Aspergillus nidulans16 reported defects Members of the protein phosphatase 1 and 2A (PP1 and in late mitosis of PP1 and PP2A mutants. However, the assays used in PP2A) families can dephosphorylate Cdk1 substrates in these studies were not specific for mitotic exit because they scored pro- biochemical extracts during mitotic exit5,6, but how this relates metaphase arrest or anaphase chromosome bridges, which can result to postmitotic reassembly of interphase structures in intact from defects in early mitosis. cells is not known. Here, we use a live-cell imaging assay and Intracellular targeting of Ser/Thr phosphatase complexes to specific RNAi knockdown to screen a genome-wide library of protein substrates is mediated by a diverse range of regulatory and targeting phosphatases for mitotic exit functions in human cells. We subunits that associate with a small group of catalytic subunits3,4,17. -

The Phosphatase JKAP/DUSP22 Inhibits T-Cell Receptor Signalling and Autoimmunity by Inactivating Lck
ARTICLE Received 8 Aug 2013 | Accepted 11 Mar 2014 | Published 9 Apr 2014 DOI: 10.1038/ncomms4618 The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck Ju-Pi Li1, Chia-Yu Yang1,Ã, Huai-Chia Chuang1,Ã, Joung-Liang Lan2,3,4,Ã, Der-Yuan Chen2,5,Ã, Yi-Ming Chen2,5,Ã, Xiaohong Wang6, Alice J. Chen6,7, John W. Belmont7 & Tse-Hua Tan1,6 JNK pathway-associated phosphatase (JKAP, also known as DUSP22 or JSP-1) is a JNK activator. The in vivo role of JKAP in immune regulation remains unclear. Here we report that JKAP directly inactivates Lck by dephosphorylating tyrosine-394 residue during T-cell receptor (TCR) signalling. JKAP-knockout T cells display enhanced cell proliferation and cytokine production. JKAP-knockout mice show enhanced T-cell-mediated immune responses and are more susceptible to experimental autoimmune encephalomyelitis (EAE). In addition, the recipient mice that are adoptively transferred with JKAP-knockout T cells show exacerbated EAE symptoms. Aged JKAP-knockout mice spontaneously develop inflammation and autoimmunity. Thus, our results indicate that JKAP is an important phosphatase that inactivates Lck in the TCR signalling turn-off stage, leading to suppression of T-cell-mediated immunity and autoimmunity. 1 Immunology Research Center, National Health Research Institutes, Zhunan 35053, Taiwan. 2 Division of Allergy, Immunology, and Rheumatology, Taichung Veterans General Hospital, Taichung 40705, Taiwan. 3 Department of Medicine, China Medical University, Taichung 40402, Taiwan. 4 Division of Rheumatology & Immunology, Department of Internal Medicine, China Medical University Hospital, Taichung 40402, Taiwan. 5 Faculty of Medicine, National Yang-Ming University, Taipei 11221, Taiwan. -
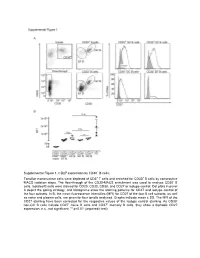
Supplemental Figure 1. CD27 Expression by CD30+ B Cells
Supplemental Figure 1. CD27 expression by CD30+ B cells. Tonsillar mononuclear cells were depleted of CD3+ T cells and enriched for CD30+ B cells by consecutive MACS isolation steps. The flow-through of the CD30-MACS enrichment was used to analyze CD30- B cells. Isolated B cells were stained for CD20, CD30, CD38, and CD27 or isotype control. Dot plots in panel A depict the gating strategy, and histograms show the staining patterns for CD27 and isotype control of the four subsets. In B, the mean fluorescence intensities (MFI) for CD27 of the four B cell subsets, as well as naive and plasma cells, are given for four tonsils analyzed. Graphs indicate mean ± SD. The MFI of the CD27 staining have been corrected for the respective values of the isotype control staining. As CD30- non-GC B cells include CD27- naïve B cells and CD27+ memory B cells, they show a biphasic CD27 expression. n.s., not significant; ** p<0.01 (unpaired t test). Supplemental Figure 2. Interleukin receptor expression by CD30+ B cells and relatedness of CD30+ EF B cells to CD21low B cells. mRNA and surface protein expression of IL2RB (A), IL21R (B) and CD21 (C) are shown for indicated B cell subsets of five tonsils (conventional (conv.) CD30- GC (germinal center) B cells (CD20highCD38+), CD30+ GC B cells, CD30- memory and CD30+ EF (extra follicular) B cells (CD20+CD38-/lowCD27+), and plasma cells (PC) (CD20+CD38high). mRNA data originate from Affymetrix genechip analyses (Tiacci et al., Blood, 2012; 120:4609-4620) and were analyzed using an unpaired t test. -

Phosphatases Page 1
Phosphatases esiRNA ID Gene Name Gene Description Ensembl ID HU-05948-1 ACP1 acid phosphatase 1, soluble ENSG00000143727 HU-01870-1 ACP2 acid phosphatase 2, lysosomal ENSG00000134575 HU-05292-1 ACP5 acid phosphatase 5, tartrate resistant ENSG00000102575 HU-02655-1 ACP6 acid phosphatase 6, lysophosphatidic ENSG00000162836 HU-13465-1 ACPL2 acid phosphatase-like 2 ENSG00000155893 HU-06716-1 ACPP acid phosphatase, prostate ENSG00000014257 HU-15218-1 ACPT acid phosphatase, testicular ENSG00000142513 HU-09496-1 ACYP1 acylphosphatase 1, erythrocyte (common) type ENSG00000119640 HU-04746-1 ALPL alkaline phosphatase, liver ENSG00000162551 HU-14729-1 ALPP alkaline phosphatase, placental ENSG00000163283 HU-14729-1 ALPP alkaline phosphatase, placental ENSG00000163283 HU-14729-1 ALPPL2 alkaline phosphatase, placental-like 2 ENSG00000163286 HU-07767-1 BPGM 2,3-bisphosphoglycerate mutase ENSG00000172331 HU-06476-1 BPNT1 3'(2'), 5'-bisphosphate nucleotidase 1 ENSG00000162813 HU-09086-1 CANT1 calcium activated nucleotidase 1 ENSG00000171302 HU-03115-1 CCDC155 coiled-coil domain containing 155 ENSG00000161609 HU-09022-1 CDC14A CDC14 cell division cycle 14 homolog A (S. cerevisiae) ENSG00000079335 HU-11533-1 CDC14B CDC14 cell division cycle 14 homolog B (S. cerevisiae) ENSG00000081377 HU-06323-1 CDC25A cell division cycle 25 homolog A (S. pombe) ENSG00000164045 HU-07288-1 CDC25B cell division cycle 25 homolog B (S. pombe) ENSG00000101224 HU-06033-1 CDKN3 cyclin-dependent kinase inhibitor 3 ENSG00000100526 HU-02274-1 CTDSP1 CTD (carboxy-terminal domain,