Partial Covariance Two-Dimensional Mass Spectrometry for Determination of Biomolecular Primary Structure
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polyphenolic Compounds Extracted and Purified from Buddleja Globosa
molecules Article Polyphenolic Compounds Extracted and Purified from Buddleja Globosa Hope (Buddlejaceae) Leaves Using Natural Deep Eutectic Solvents and Centrifugal Partition Chromatography Jeniffer Torres-Vega 1 , Sergio Gómez-Alonso 2 , José Pérez-Navarro 2 , Julio Alarcón-Enos 3 and Edgar Pastene-Navarrete 1,3,* 1 Laboratorio de Farmacognosia, Departamento de Farmacia, Facultad de Farmacia, Universidad de Concepción, Concepción PC4030000, Chile; [email protected] 2 Regional Institute for Applied Scientific Research, Faculty of Chemical Sciences, University of Castilla-La Mancha, PC13071 Castilla-La Mancha, Spain; [email protected] (S.G.-A.); [email protected] (J.P.-N.) 3 Laboratorio de Síntesis y Biotransformación de Productos Naturales, Universidad del Bío-Bío, Chillán PC3800708, Chile; [email protected] * Correspondence: [email protected]; Tel.: +56-(42)-246-3000 Abstract: Chemical profiling of Buddleja globosa was performed by high-performance liquid chro- matography coupled to electrospray ionization (HPLC-DAD-ESI-IT/MS) and quadrupole time-of-flight high-resolution mass spectrometry (HPLC-ESI-QTOF/MS). The identification of 17 main phenolic com- pounds in B. globosa leaf extracts was achieved. Along with caffeoyl glucoside isomers, caffeoylshikimic Citation: Torres-Vega, J.; acid and several verbascoside derivatives (β-hydroxyverbascoside and β-hydroxyisoverbascoside) were Gómez-Alonso, S.; Pérez-Navarro, J.; Alarcón-Enos, J.; Pastene-Navarrete, identified. Among flavonoid compounds, the presence of 6-hydroxyluteolin-7-O-glucoside, quercetin-3- E. Polyphenolic Compounds O-glucoside, luteolin 7-O-glucoside, apigenin 7-O-glucoside was confirmed. Campneoside I, forsytho- Extracted and Purified from Buddleja side B, lipedoside A and forsythoside A were identified along with verbascoside, isoverbascoside, Globosa Hope (Buddlejaceae) Leaves eukovoside and martynoside. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -
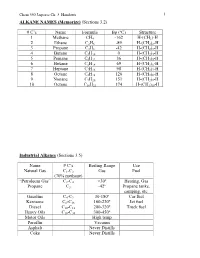
C's Name Formula Bp (ºC) Structure 1 Methane CH4 -162 H-(CH2)
Chem 350 Jasperse Ch. 3 Handouts 1 ALKANE NAMES (Memorize) (Sections 3.2) # C’s Name Formula Bp (ºC) Structure 1 Methane CH4 -162 H-(CH2)-H 2 Ethane C2H6 -89 H-(CH2)2-H 3 Propane C3H8 -42 H-(CH2)3-H 4 Butane C4H10 0 H-(CH2)4-H 5 Pentane C5H12 36 H-(CH2)5-H 6 Hexane C6H14 69 H-(CH2)6-H 7 Heptane C7H16 98 H-(CH2)7-H 8 Octane C8H18 126 H-(CH2)8-H 9 Nonane C9H20 151 H-(CH2)9-H 10 Octane C10H22 174 H-(CH2)10-H Industrial Alkanes (Sections 3.5) Name # C’s Boiling Range Use Natural Gas C1-C3 Gas Fuel (70% methane) “Petroleum Gas” C2-C4 <30º Heating, Gas Propane C3 -42º Propane tanks, camping, etc. Gasoline C4-C9 30-180º Car fuel Kerosene C8-C16 160-230º Jet fuel Diesel C10-C18 200-320º Truck fuel Heavy Oils C16-C30 300-450º Motor Oils High temp Paraffin Vacuum Asphalt Never Distills Coke Never Distills Chem 350 Jasperse Ch. 3 Handouts 2 Nomenclature of Alkanes (Sections 3.3) Systematic IUPAC Rules for Branched and Substituted Alkanes (Section 3.3B) 1. Longest continuous C-chain “core name” 2. Number core chain from an end nearest a substituent 3. Name substituents as “alkyl” groups: 4. Specify the location of substituents using numbers (hyphenate the #’s) • If >2 substituents, list alphabetically • Use di-, tri-, tetra- if the same substituent is repeated. (But ignore these in alphabetizing). Punctuation Notes: • Hyphenate numbers • Do not put a space between substituents and the core name Special Names for Some 3 or 4-carbon Substituents H3C CH3 CH Memorize H3C C H3C CH3 Isopropyl t-butyl or tert-butyl H2 H2 CH3 H3C C H3C C CH H3C CH Others C C C C CH3 H3C C H2 H H H2 2 2 H2 n-propyl n-butyl isobutyl s-butyl (n for "normal") Another Classification System Primary (1º): with one attached carbon Secondary (2º): with two attached carbons Tertiary (3º): with three attached carbons H C C C C C C C C H 1º H 2º C 3º Very Complex Substituents (Not responsible) Substituent: (1-ethyl-2,3-dimethylpentyl) Overall: 9-(1-ethyl-2,3-dimethylpentyl)nonadecane Chem 350 Jasperse Ch. -

Influence of Conjugation Axis on the Optical and Electronic Properties Of
Article pubs.acs.org/joc Influence of Conjugation Axis on the Optical and Electronic Properties of Aryl-Substituted Benzobisoxazoles † ‡ ‡ † † Brian C. Tlach, Aimeé L. Tomlinson, Alden G. Ryno, Dawn D. Knoble, Dana L. Drochner, † Kyle J. Krager, and Malika Jeffries-EL*, † Department of Chemistry, Iowa State University, Ames, Iowa 50010, United States ‡ Department of Chemistry, University of North Georgia, Dahlonega, Georgia 30597, United States *S Supporting Information ABSTRACT: Six different 2,6-diethyl-4,8-diarylbenzo[1,2-d:4,5-d′]bis(oxazoles) and four different 2,4,6,8-tetraarylbenzobisoxazoles were synthesized in two steps: a Lewis acid catalyzed orthoester cyclization followed by a Suzuki or Stille cross-coupling with various arenes. The influence of aryl group substitution and/or conjugation axis variation on the optical and electronic properties of these benzobis(oxazole) (BBO) compounds was evaluated. Structural modifications could be used to alter the HOMO, LUMO, and band gap over a range of 1.0, 0.5, and 0.5 eV, respectively. However, depending on the location and identity of the substituent, the HOMO level can be altered without significantly impacting the LUMO level. This is supported by the calculated frontier molecular orbitals. Our results indicate that the FMOs and band gaps of benzobisoxazoles can be readily modified either jointly or individually. ■ INTRODUCTION Among the aforementioned examples, the benzo[1,2-d:4,5- d′]bis(oxazole) (BBO)-based cruciforms are particularly During the past four decades, interest in the development of π- interesting, since these molecules have two different con- conjugated materials has increased due to their potential use as jugation pathways: 2,6-conjugation through the oxazole rings replacements for inorganic materials in a variety of semi- and 4,8-conjugation through the central benzene ring (Scheme conducting applications including field effect transistors − − 1). -

Physical Sciences Content
Foreword In order to improve learning outcomes the Department of Basic Education conducted research to determine the specific areas that learners struggle with in Grade 12 examinations. The research included a trend analysis by subject experts of learner performance over a period of five years as well as learner examination scripts in order to diagnose deficiencies or misconceptions in particular content areas. In addition, expert teachers were interviewed to determine the best practicesto ensure mastery of thetopic by learners and improve outcomes in terms of quality and quantity. The results of the research formed the foundation and guiding principles for the development of the booklets. In each identified subject, key content areas were identified for the development of material that will significantly improve learner's conceptual understanding whilst leading to improved performance in the subject. The booklets are developed as part of a series of booklets, with each bookletfocussing onlyon one specific challenging topic. The selected content is explained in detail and include relevant concepts from Grades 10 - 12 to ensure conceptual understanding. The main purpose of these booklets is to assist learners to master the content starting from a basic conceptual level of understanding to the more advanced level. The content in each booklets is presented in an easy to understand manner including the use of mind maps, summaries and exercises to support understanding and conceptual progression. These booklets should ideally be used as part of a focussed revision or enrichment program by learners after the topics have been taught in class. The booklets encourage learners to take ownership of their own learning and focus on developing and mastery critical content and skills such as reading and higher order thinking skills. -

Heuristics for Searching Chemical Structures a Thesis Presented To
Heuristics for Searching Chemical Structures A Thesis presented to the Faculty of the Graduate School University of Missouri-Columbia In Partial Fulfillment of the Requirements for the Degree Master of Science by Nandini Basu Dr. Toni Kazic, Thesis Supervisor May 2007 The undersigned, appointed by the Dean of the Graduate School, have examined the thesis entitled HEURISTICS FOR SEARCHING CHEMICAL STRUCTURES presented by Nandini Basu A candidate for the degree of Master of Science And hereby certify that in their opinion it is worthy of acceptance. Dr. Toni Kazic Dr. Chi-Ren Shyu Dr. Mary Schaeffer ACKNOWLEDGMENTS I am deeply indebted to my research advisor Dr. Toni Kazic for giving me an opportunity to resume my research in and complete my Computer Science MS program. Without her guidance, support and encouragement this would not have been possible. I would like to thank Dr. Mary Polaco and Dr. Shyu for serving in my committee and helping me in my thesis. I would like to thank the Klotho team for helping me with structures. And also the other team members Amith, Archana, Avanthi, Bill, Deepthi, Gaurav, Raman, Jiahui, Fatten, Meeta and Michael. My friends made a difficult journey lot of fun and much easier, especially Cathy, Charu, Deepak, Karan, Karthik, Paco, Nikunj, Raj, Shiva, Shraddha, Sundeep and Vibha. All my relatives gave me constant encouragement and I would like to take this opportunity to thank them. And above all, I would like to thank my father Prabhat Kumar Basu, my mother Minoti Basu, my brother Pradipta Kumar Basu and my sister-in-law Sarmistha Basu. -

1 Pesticide Analysis by Mass Spectrometry Purpose
Pesticide Analysis by Mass Spectrometry Purpose: The purpose of this assignment is to introduce concepts of mass spectrometry (MS) as they pertain to the qualitative and quantitative analysis of organochlorine pesticides found in water samples. Learning Outcomes At the end of this assignment, students will be able to: 1. Explain the processes involved in the ionization of compounds in GCMS. 2. Predict isotopic distributions and identify chlorinated compounds by their MS isotopic signature and fragmentation patterns. 3. Identify and use unique ions for MS quantitation in complex samples. 4. Using a sampling scheme for Lake Nakuru, determine the concentrations of DDT from GCMS results and draw some conclusions as to whether the levels of DDT detected in Lake Nakuru water play a role in flamingos’ death. In the previous modules you have been introduced to the problem of flamingo die off events in Lake Nakuru, and the analytical chemistry considerations needed to address this problem including sampling, sample preparation, and gas chromatography. In addition to this discussion of MS analysis, the following module on validation of the analytical method addresses issues related to ensuring that the results obtained are of sufficient quality that they can be used to draw appropriate scientific conclusions. In this section, aspects of ionization and mass analysis are explored and example GCMS results are presented. Introduction This section is designed to provide an introduction to mass spectrometry (MS) in the context of its utilization as a gas chromatography detector. The basic concepts of ionization, mass analysis, isotope ratios, and mass spectral interpretation are introduced. -

Chapter 24 "Organic Compounds"
This is “Organic Compounds”, chapter 24 from the book Principles of General Chemistry (index.html) (v. 1.0). This book is licensed under a Creative Commons by-nc-sa 3.0 (http://creativecommons.org/licenses/by-nc-sa/ 3.0/) license. See the license for more details, but that basically means you can share this book as long as you credit the author (but see below), don't make money from it, and do make it available to everyone else under the same terms. This content was accessible as of December 29, 2012, and it was downloaded then by Andy Schmitz (http://lardbucket.org) in an effort to preserve the availability of this book. Normally, the author and publisher would be credited here. However, the publisher has asked for the customary Creative Commons attribution to the original publisher, authors, title, and book URI to be removed. Additionally, per the publisher's request, their name has been removed in some passages. More information is available on this project's attribution page (http://2012books.lardbucket.org/attribution.html?utm_source=header). For more information on the source of this book, or why it is available for free, please see the project's home page (http://2012books.lardbucket.org/). You can browse or download additional books there. i Chapter 24 Organic Compounds In Chapter 2 "Molecules, Ions, and Chemical Formulas", you were introduced to the major classes of organic compounds, covalent compounds composed primarily of carbon and hydrogen. Organic substances have been used throughout this text to illustrate the differences between ionic and covalent bonding and to demonstrate the intimate connection between the structures of compounds and their chemical reactivity. -

Chapter 21 Organic Chemistry
CHAPTER 21 ORGANIC CHEMISTRY Hydrocarbons 1. A hydrocarbon is a compound composed of only carbon and hydrogen. A saturated hydro- carbon has only carbon-carbon single bonds in the molecule. An unsaturated hydrocarbon has one or more carbon-carbon multiple bonds but may also contain carbon-carbon single bonds. A normal hydrocarbon has one chain of consecutively bonded carbon atoms. A branched hydrocarbon has at least one carbon atom not bonded to the end carbon of a chain of consecutively bonded carbon atoms. Instead, at least one carbon atom forms a bond to an inner carbon atom in the chain of consecutively bonded carbon atoms. 2. To determine the number of hydrogens bonded to the carbons in cyclic alkanes (or any alkane where they may have been omitted), just remember that each carbon has four bonds. In cycloalkanes, only the C−C bonds are shown. It is assumed you know that the remaining bonds on each carbon are C−H bonds. The number of C−H bonds is that number required to give the carbon four total bonds. 3. In order to form, cyclopropane and cyclobutane are forced to form bond angles much smaller than the preferred 109.5° bond angles. Cyclopropane and cyclobutane easily react in order to obtain the preferred 109.5° bond angles. 4. Aromatic hydrocarbons are a special class of unsaturated hydrocarbons based on the benzene ring. Benzene has the formula C6H6. It is a planar molecule (all atoms are in the same plane). Each carbon in benzene is attached to three other atoms; it exhibits trigonal planar geometry with 120° bond angles. -

A New Derivation of Structural Isomers of Straight Chain Alkanes
Int'l Conference on Advanced Computational Technologies & Creative Media (ICACTCM’2014) Aug. 14-15, 2014 Pattaya (Thailand) A New Derivation of Structural Isomers of Straight Chain Alkanes Alfred Addo-Yobo odd carbon number straight chain alkanes where „„n‟‟ is the Abstract—Structural isomers are very important due to their number of carbon atoms of straight chain alkane. different properties. The structural isomers of light weighted straight chain alkanes such as pentane, hexane and butane are easy to derive, Structural isomers usually come with certain numbers as they contain fewer number of carbon atoms. With easy substitution preceding the substituents which are known as locant numbers of one or more hydrogen atoms with one or several alkyl groups on specific carbons, different forms of the same compound can be as stated in Wumbee Books, pg. 105. The attached numbers determined. This conventional module of alkyl and hydrogen groups which show the position of substituents on a specific carbon substitution to derive structural isomers, though efficient, may require atom may appear as single, double, triple or any number of thorough check up of substituent positions on specific carbons to number combinations. ensure that, non-permissible structures are not listed. This procedure Examples of single locant number structural isomers are 2 gets cumbersome when dealing with straight chain alkanes of higher methyl pentane, 3 methyl pentane. Double locant number carbon numbers. In this paper, we introduce a new technique by which all possible structural isomers of any straight chain alkane structural isomers could be 2, 3 dimethyl hexane, 3, 4 whatsoever can be derived, by a number combination theory as well dimethyl octane or sometimes triple numbers such as 2, 3, 4 as introduce three new postulates that validates a compound as being trimethyl nonane or 2, 2, 5 trimethyl decane. -

Arene Capsules in the Solid State
Organic Chemistry Frontiers 2H NMR Reveals Liquid State-Like Dynamics of Arene Guests Inside Hexameric Pyrogallol[4]arene Capsules in the Solid State Journal: Organic Chemistry Frontiers Manuscript ID QO-RES-02-2019-000232.R1 Article Type: Research Article Date Submitted by the 22-Mar-2019 Author: Complete List of Authors: Islas, Irazema; San Diego State University, Department of Chemistry and Biochemistry Stengel, Dillan; San Diego State University, Department of Chemistry and Biochemistry Garcia, Cesar; San Diego State University, Department of Chemistry and Biochemistry Addison, J; San Diego State University, Department of Chemistry and Biochemistry Samaan, George; San Diego State University, Department of Chemistry and Biochemistry Holland, Gregorgy; San Diego State University, Department of Chemistry and Biochemistry Purse, Byron W; San Diego State University, Department of Chemistry and Biochemistry Page 1 of 6 PleaseOrganic do Chemistry not adjust Frontiers margins 1 2 3 4 Journal Name 5 6 7 8 ARTICLE 9 10 11 2 12 H NMR Reveals Liquid State-Like Dynamics of Arene Guests 13 Inside Hexameric Pyrogallol[4]arene Capsules in the Solid State.† 14 Received 00th January 20xx, a a a a a 15 Accepted 00th January 20xx Irazema J. Islas,‡ Dillan Stengel,‡ Cesar A. Garcia, J. Bennett Addison, George N. Samaan, 16 Gregory P. Holland,a and Byron W. Purse*a,b 17 DOI: 10.1039/x0xx00000x 18 www.rsc.org/ The dynamics of guests in molecular encapsulation complexes have been studied extensively in solution, but the 19 corresponding behavior of those guests when the capsules are present in the solid state are not as well understood. -

Identify Organic Functional Groups, Bond Types And
Objective 1: Identify organic functional groups, bond types and IR, draw skeletal structures, and distinguish between the same compound, isomers, different compounds, and resonance structures. Quiz Practice problems: Key ideas: Most substances are organic compounds. Biomolecules are organic compounds. Organic compounds contain C, H, O, N, S, P, X Organic compounds are classified by functional groups – a small group of 2 to 5 atoms within an organic compound that has specific properties. Organic compounds are represented by Lewis structures. General bonding rules: C = 4 bonds + 0 lone pairs, N = 3 bonds + 1 lone pairs, O = 2 bonds + 2 lone pairs, X = 1 bonds + 3 lone pairs, H = 1 bonds + 0 lone pairs. In a Lewis structure, a covalent bond shows electrons shared between 2 atoms only --> localized electrons. But electrons can be shared between 3 or more atoms – delocalized electrons. Need to show resonance structures to represent delocalized electrons. Experiments show electrons are delocalized – see infrared (IR) spectroscopy. Organic compounds can be large – use skeletal structure as a shortcut for a Lewis structure. Organic compounds can be large – there are different ways to connect (bond) atoms in a compound. Two compounds that have the same chemical formula but different connectivity (bonding) are called isomers. When a substance is exposed to infrared (IR) radiation, bonds between atoms vibrate (stretch or bend). IR spectroscopy is used to determine bond types, e.g., C-H, O-H, in a compound. IR spectroscopy is used to help determine structure of a molecule. Skills: Given Lewis structure, draw skeletal structure and vice versa. Draw isomers of a compound.