Spectroscopic Characterization of Structural Isomers of Naphthalene: 1-Phenyl-1-Butyn-3-Ene ⇑ Joshua A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rotational Spectroscopy
DAV University Jalandhar Department of Physics Study Material: PHY 512 Atomic & Molecular Spectroscopy M.Sc (Hons.) Physics 2nd sem Source : internet -http://www.phys.ubbcluj.ro/~dana.maniu/OS/BS_3.pdf BOOK: Fundamentals of Molecular Spectroscopy CN Banwell Mc Graw Hill Rotational spectroscopy - Involve transitions between rotational states of the molecules (gaseous state!) - Energy difference between rotational levels of molecules has the same order of magnitude with microwave energy - Rotational spectroscopy is called pure rotational spectroscopy, to distinguish it from roto-vibrational spectroscopy (the molecule changes its state of vibration and rotation simultaneously) and vibronic spectroscopy (the molecule changes its electronic state and vibrational state simultaneously) Molecules do not rotate around an arbitrary axis! Generally, the rotation is around the mass center of the molecule. The rotational axis must allow the conservation of M R α pα const kinetic angular momentum. α Rotational spectroscopy Rotation of diatomic molecule - Classical description Diatomic molecule = a system formed by 2 different masses linked together with a rigid connector (rigid rotor = the bond length is assumed to be fixed!). The system rotation around the mass center is equivalent with the rotation of a particle with the mass μ (reduced mass) around the center of mass. 2 2 2 2 m1m2 2 The moment of inertia: I miri m1r1 m2r2 R R i m1 m2 Moment of inertia (I) is the rotational equivalent of mass (m). Angular velocity () is the equivalent -

Reliable Structures and Energetics for Two New Delocalized Pбббp
PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics Reliable structures and energetics for two new delocalized pÁÁÁp prototypes: cyanogen dimer and diacetylene dimerwz Brian W. Hopkins, Adel M. ElSohly and Gregory S. Tschumper* Received 22nd November 2006, Accepted 3rd January 2007 First published as an Advance Article on the web 7th February 2007 DOI: 10.1039/b616878g Two new prototype delocalized pÁÁÁp complexes are introduced: the dimers of cyanogen, (NRC–CRN)2, and diacetylene, (HCRC–CRCH)2. These dimers have properties similar to larger delocalized pÁÁÁp systems such as benzene dimer but are small enough that they can be probed in far greater detail with high accuracy electronic structure methods. Parallel-slipped and T-shaped structures of both cyanogen dimer and diacetylene dimer have been optimized with 15 different procedures. The effects of basis set size, theoretical method, counterpoise correction, and the rigid monomer approximation on the structure and energetics of each dimer have been examined. MP2 and CCSD(T) optimized geometries for all four dimer structures are reported, as well as estimates of the CCSD(T) complete basis set (CBS) interaction energy for every optimized geometry. The data reported here suggest that future optimizations of delocalized pÁÁÁp clusters should be carried out with basis sets of triple-z quality. Larger basis sets and the expensive counterpoise correction to the molecular geometry are not necessary. The rigid monomer approximation has very little effect on structure and energetics of these dimers and may be used without consequence. Due to a consistent cancellation of errors, optimization with the MP2 method leads to CCSD(T)/CBS interaction energies that are within 0.2 kcal molÀ1 of those for structures optimized with the CCSD(T) method. -

The Synthesis of a Polydiacetylene to Create a Novel Sensory Material
SELDE, KRISTEN A., M.S. The Synthesis of a Polydiacetylene to Create a Novel Sensory Material. (2007) Directed by Dr. Darrell Spells. 47pp. Sensory materials that respond to chemical and mechanical stimuli are under development in many laboratories. There are many significant uses of polydiacetylene compounds as sensory material. They have been applied to drug delivery, drug design, biomolecule development, cosmetics, and national security. In this study, experiments were carried out toward the development of a novel sensory material based on the established synthetic research on polydiacetylene compounds. Synthetic routes toward sensory materials with different head groups, different carbon chains lengths, and the incorporation of molecular imprints were explored. Diacetylene moieties, which can be used for polymer vesicle formation, were prepared by two main routes. In one route, 1-iodo-1-octyne and 1-iodo-1-dodecyne were prepared as starting materials for the synthesis of two diacetylene compounds (Diacetylene I and Diacetylene II). In the other route, a mesityl alkyne was used to prepare 5-iodo-1-pentyne, which was then used to prepare a triethylamino alkyne. This in turn was used to synthesize a diacetylene (Diacetylene III). Although each diacetylene product was formed, purification by column chromatography was found to be difficult. Experiments in vesicle formation, with and without molecular imprints, were also carried out using commercially available diacetylenes . THE SYNTHESIS OF A POLYDIACETYLENE TO CREATE A NOVEL SENSORY -

Polyphenolic Compounds Extracted and Purified from Buddleja Globosa
molecules Article Polyphenolic Compounds Extracted and Purified from Buddleja Globosa Hope (Buddlejaceae) Leaves Using Natural Deep Eutectic Solvents and Centrifugal Partition Chromatography Jeniffer Torres-Vega 1 , Sergio Gómez-Alonso 2 , José Pérez-Navarro 2 , Julio Alarcón-Enos 3 and Edgar Pastene-Navarrete 1,3,* 1 Laboratorio de Farmacognosia, Departamento de Farmacia, Facultad de Farmacia, Universidad de Concepción, Concepción PC4030000, Chile; [email protected] 2 Regional Institute for Applied Scientific Research, Faculty of Chemical Sciences, University of Castilla-La Mancha, PC13071 Castilla-La Mancha, Spain; [email protected] (S.G.-A.); [email protected] (J.P.-N.) 3 Laboratorio de Síntesis y Biotransformación de Productos Naturales, Universidad del Bío-Bío, Chillán PC3800708, Chile; [email protected] * Correspondence: [email protected]; Tel.: +56-(42)-246-3000 Abstract: Chemical profiling of Buddleja globosa was performed by high-performance liquid chro- matography coupled to electrospray ionization (HPLC-DAD-ESI-IT/MS) and quadrupole time-of-flight high-resolution mass spectrometry (HPLC-ESI-QTOF/MS). The identification of 17 main phenolic com- pounds in B. globosa leaf extracts was achieved. Along with caffeoyl glucoside isomers, caffeoylshikimic Citation: Torres-Vega, J.; acid and several verbascoside derivatives (β-hydroxyverbascoside and β-hydroxyisoverbascoside) were Gómez-Alonso, S.; Pérez-Navarro, J.; Alarcón-Enos, J.; Pastene-Navarrete, identified. Among flavonoid compounds, the presence of 6-hydroxyluteolin-7-O-glucoside, quercetin-3- E. Polyphenolic Compounds O-glucoside, luteolin 7-O-glucoside, apigenin 7-O-glucoside was confirmed. Campneoside I, forsytho- Extracted and Purified from Buddleja side B, lipedoside A and forsythoside A were identified along with verbascoside, isoverbascoside, Globosa Hope (Buddlejaceae) Leaves eukovoside and martynoside. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -
C's Name Formula Bp (ºC) Structure 1 Methane CH4 -162 H-(CH2)
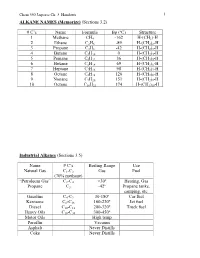
Chem 350 Jasperse Ch. 3 Handouts 1 ALKANE NAMES (Memorize) (Sections 3.2) # C’s Name Formula Bp (ºC) Structure 1 Methane CH4 -162 H-(CH2)-H 2 Ethane C2H6 -89 H-(CH2)2-H 3 Propane C3H8 -42 H-(CH2)3-H 4 Butane C4H10 0 H-(CH2)4-H 5 Pentane C5H12 36 H-(CH2)5-H 6 Hexane C6H14 69 H-(CH2)6-H 7 Heptane C7H16 98 H-(CH2)7-H 8 Octane C8H18 126 H-(CH2)8-H 9 Nonane C9H20 151 H-(CH2)9-H 10 Octane C10H22 174 H-(CH2)10-H Industrial Alkanes (Sections 3.5) Name # C’s Boiling Range Use Natural Gas C1-C3 Gas Fuel (70% methane) “Petroleum Gas” C2-C4 <30º Heating, Gas Propane C3 -42º Propane tanks, camping, etc. Gasoline C4-C9 30-180º Car fuel Kerosene C8-C16 160-230º Jet fuel Diesel C10-C18 200-320º Truck fuel Heavy Oils C16-C30 300-450º Motor Oils High temp Paraffin Vacuum Asphalt Never Distills Coke Never Distills Chem 350 Jasperse Ch. 3 Handouts 2 Nomenclature of Alkanes (Sections 3.3) Systematic IUPAC Rules for Branched and Substituted Alkanes (Section 3.3B) 1. Longest continuous C-chain “core name” 2. Number core chain from an end nearest a substituent 3. Name substituents as “alkyl” groups: 4. Specify the location of substituents using numbers (hyphenate the #’s) • If >2 substituents, list alphabetically • Use di-, tri-, tetra- if the same substituent is repeated. (But ignore these in alphabetizing). Punctuation Notes: • Hyphenate numbers • Do not put a space between substituents and the core name Special Names for Some 3 or 4-carbon Substituents H3C CH3 CH Memorize H3C C H3C CH3 Isopropyl t-butyl or tert-butyl H2 H2 CH3 H3C C H3C C CH H3C CH Others C C C C CH3 H3C C H2 H H H2 2 2 H2 n-propyl n-butyl isobutyl s-butyl (n for "normal") Another Classification System Primary (1º): with one attached carbon Secondary (2º): with two attached carbons Tertiary (3º): with three attached carbons H C C C C C C C C H 1º H 2º C 3º Very Complex Substituents (Not responsible) Substituent: (1-ethyl-2,3-dimethylpentyl) Overall: 9-(1-ethyl-2,3-dimethylpentyl)nonadecane Chem 350 Jasperse Ch. -

Guidance Document Peroxide-Forming Chemicals
Guidance Document Peroxide-Forming Chemicals Some chemicals can form peroxides under normal storage conditions. Some of the peroxide chemicals are unstable, especially when dried or concentrated, and can explode violently when subjected to heat, light or mechanical shock. In addition, some of the inadvertently formed peroxides can initiate other unexpected violent reactions (e.g. polymerizations) with other chemicals. When possible and practical for your work, purchase chemicals that have inhibitors added by the manufacturer. Label peroxide-forming chemicals with date received and date opened. Store peroxide-formers in airtight opaque containers with screw caps. Consider oxygen exclusion methods such as purging with inert gas or sealing containers with parafilm. Inspect containers for signs of peroxide formation. Do not open a container which has crystals or a visible cloudiness. Call EHS to come remove it. The friction caused by opening a lid can cause an explosion. Liquids can be tested for presence of peroxide. This is especially important prior to distilation. Most explosions of peroxide forming chemicals occur when a material is distilled to dryness. Peroxide test kits are available from chemical vendors. Contact EHS for additional guidance. Classification Table for Peroxide-Forming Chemicals Class I:: Unsaturated materials, especially those of low molecular weight, may polymerize violently and hazardously due to peroxide initiation. These chemicals can spontaneously decompose, becoming explosive after exposure to air with concentration. Discard unopened containers within 3 months. Opened containers should be tested for peroxides every 2 months. Acrylic acid Tetrafluoroethylene Acrylonitrile Vinyl acetate 1,3-Butadiene Vinyl acetylene Chlorobutadiene (chloroprene) Vinyl chloride Chlorotrifluoroethylene Vinyl pyridine Methyl methacrylate Vinylidiene chloride Styrene Class II: The following chemicals are a peroxide hazard upon concentration (distillation/evaporation). -

Chemical Reactivity of Naphthalenecarboxylate-Protected
Article pubs.acs.org/JPCC Chemical Reactivity of Naphthalenecarboxylate-Protected Ruthenium Nanoparticles: Intraparticle Charge Delocalization Derived from Interfacial Decarboxylation † † † ‡ ‡ † ‡ Limei Chen, Peiguang Hu, Christopher P. Deming, Wei Li, Ligui Li, and Shaowei Chen*, , † Department of Chemistry and Biochemistry, University of California, 1156 High Street, Santa Cruz, California 95064, United States ‡ New Energy Research Institute, School of Environment and Energy, South China University of Technology, Guangzhou Higher Education Mega Center, Guangzhou 510006, China ABSTRACT: Ruthenium nanoparticles were prepared by thermolytic reduction of RuCl3 in 1,2-propanediol containing sodium 2-naphthalenecarboxylate. Transmission electron microscopic measurements showed that the average diameter of the resulting 2- naphthalenecarboxylate-protected ruthenium nanoparticles (RuCOONA) was 1.30 ± 0.27 nm. Interestingly, hydrothermal treatment of the nanoparticles at controlled temperatures led to decarboxylation at the metal−ligand interface, and the naphthalenyl moieties became directly bonded to the metal cores, which was confirmed by infrared and X-ray photoelectron spectroscopic measurements. In comparison with the as-produced RuCOONA nanoparticles, the decarboxylated nanoparticles (RuNA) exhibited markedly different optical and electronic properties, as manifested by an apparent red shift of the photoluminescence profiles, which was ascribed to electronic coupling between the particle-bound naphthalene groups. Electrochemical measurements exhibited consistent results where a negative shift was observed of the formal potential of the particle-bound naphthalene moieties. This was attributed to intraparticle charge delocalization that led to extended spilling of nanoparticle core electrons to the naphthalene moieties. ■ INTRODUCTION Conjugated metal−ligand interfacial bonds may also be produced by exploiting the unique interfacial reactivity of Organically capped metal nanoparticles have been attracting organic ligands on nanoparticle surfaces. -

Influence of Conjugation Axis on the Optical and Electronic Properties Of
Article pubs.acs.org/joc Influence of Conjugation Axis on the Optical and Electronic Properties of Aryl-Substituted Benzobisoxazoles † ‡ ‡ † † Brian C. Tlach, Aimeé L. Tomlinson, Alden G. Ryno, Dawn D. Knoble, Dana L. Drochner, † Kyle J. Krager, and Malika Jeffries-EL*, † Department of Chemistry, Iowa State University, Ames, Iowa 50010, United States ‡ Department of Chemistry, University of North Georgia, Dahlonega, Georgia 30597, United States *S Supporting Information ABSTRACT: Six different 2,6-diethyl-4,8-diarylbenzo[1,2-d:4,5-d′]bis(oxazoles) and four different 2,4,6,8-tetraarylbenzobisoxazoles were synthesized in two steps: a Lewis acid catalyzed orthoester cyclization followed by a Suzuki or Stille cross-coupling with various arenes. The influence of aryl group substitution and/or conjugation axis variation on the optical and electronic properties of these benzobis(oxazole) (BBO) compounds was evaluated. Structural modifications could be used to alter the HOMO, LUMO, and band gap over a range of 1.0, 0.5, and 0.5 eV, respectively. However, depending on the location and identity of the substituent, the HOMO level can be altered without significantly impacting the LUMO level. This is supported by the calculated frontier molecular orbitals. Our results indicate that the FMOs and band gaps of benzobisoxazoles can be readily modified either jointly or individually. ■ INTRODUCTION Among the aforementioned examples, the benzo[1,2-d:4,5- d′]bis(oxazole) (BBO)-based cruciforms are particularly During the past four decades, interest in the development of π- interesting, since these molecules have two different con- conjugated materials has increased due to their potential use as jugation pathways: 2,6-conjugation through the oxazole rings replacements for inorganic materials in a variety of semi- and 4,8-conjugation through the central benzene ring (Scheme conducting applications including field effect transistors − − 1). -

Chromatic Studies of a Polymerizable Diacetylene Hydrogen Bonding Self-Assembly: a “Self-Folding” Process to Explain the Chromatic Changes of Polydiacetylenes
3972 Langmuir 1999, 15, 3972-3980 Chromatic Studies of a Polymerizable Diacetylene Hydrogen Bonding Self-Assembly: A “Self-Folding” Process To Explain the Chromatic Changes of Polydiacetylenes Qun Huo, K. C. Russell,† and Roger M. Leblanc* Center for Supramolecular Science, Department of Chemistry, University of Miami, P.O. Box 249118, Coral Gables, Florida 33124 Received January 11, 1999. In Final Form: March 9, 1999 In the present study, a new diacetylene compound (PDATAZ), which readily forms a complementary hydrogen bonding self-assembly at the air-water interface or in the solid state with barbituric acid (BA) or cyanuric acid (CA), was designed and synthesized. The photopolymerization studies of PDATAZ and its assembly with BA or CA have revealed some important insights on the chromatic properties of polydiacetylenes. It was found that the chromatic property of polydiacetylenes is determined by whether the polymer chain is capable of adopting a linear chainlike shape. With the continuous increase of the length of the polymer chain, the original linear polyenyne backbone starts to “self-fold” to a “zigzag” structure due to the free rotation of single bonds within the polymer chain. The efficient π-electron delocalization along the polyenyne backbone is interrupted by this process, leading to a chromatic change from the blue to red form of polydiacetylenes. If there are strong intermolecular interactions existing between the polar groups of the side chains, such as the complementary hydrogen bonding network between the triaminotriazine (TAZ) moiety of the diacetylene amphiphile and its complementary components, the movement of the side chains is restricted and the folding process of the polymer backbone is inhibited. -

Molecular Design and Self-Assembly of Polydiacetylene for Biosensors and Sensor Arrays
Molecular Design and Self-assembly of Polydiacetylene for Biosensors and Sensor Arrays by Ji Seok Lee A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Macromolecular Science and Engineering) in The University of Michigan 2011 Doctoral Committee: Professor Jinsang Kim, Chair Professor L. Jay Guo Professor Kenichi Kuroda Professor David C. Martin Jiseok Lee © 2011 All Rights Reserved ACKNOWLEDGMENTS I would like to thank my advisor, Professor Jinsang Kim. He guided me with a great patience for my experimental kowledge as well as my presentation skill improvement. He has always encouraged me even when I made mistakes. I would like to give my sincere respect to Prof Kim for his generous concern. I could not have published good journals, enjoyed the research and successfully finished the doctoral program without his advice. I also would like to thank my committee members, Prof Martin, Prof Kuroda and Prof Guo. Under their guidance, I could draw a big picture of my research and train myself as a scientist not a technician. It was a great honor for me to do my research under their thoughtful guidance. I also thanks to Prof Laine and Nonna. I could finish my degree as a Macro student by his permission. Also Nonna helped me a lot for every student business. She saved me a lot of times from troubles. I would like to thank all of my lab members. It was good experience for me to work with them. The past members of the Kim’s group, Dr Kim and Dr Jo gave me a lot of advice. -

UC Santa Barbara Dissertation Template
UNIVERSITY OF CALIFORNIA Santa Barbara Laser Spectroscopy and Photodynamics of Alternative Nucleobases and Organic Dyes A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Chemistry by Jacob Alan Berenbeim Committee in charge: Professor Mattanjah de Vries, Chair Professor Steve Buratto Professor Michael Gordon Professor Martin Moskovits December 2017 The dissertation of Jacob Alan Berenbeim is approved. ____________________________________________ Steve Buratto ____________________________________________ Michael Gordon ____________________________________________ Martin Moskovits ____________________________________________ Mattanjah de Vries, Committee Chair October 2017 Laser Spectroscopy and Photodynamics of Alternative Nucleobases and Organic Dyes Copyright © 2017 by Jacob Alan Berenbeim iii ACKNOWLEDGEMENTS To my wife Amy thank you for your endless support and for inspiring me to match your own relentless drive towards reaching our goals. To my parents and my brothers Eli and Gabe thank you for your love and visits to Santa Barbara, CA. To my advisor Mattanjah and my lab mates thank you for the incredible opportunity to share ideas and play puppets with the fabric of space. And to my cat Lola, you’re a good cat. iv VITA OF JACOB ALAN BERENBEIM October 2017 EDUCATION University of California, Santa Barbara CA Fall 2017 PhD, Physical Chemistry Advisor: Prof. Mattanjah S. de Vries University of Puget Sound, Tacoma WA 2009 BS, Chemistry Advisor: Prof. Daniel Burgard LABORATORY TECHNIQUES Photophysics by UV/VIS and IR pulsed laser spectroscopy, optical alignment, oa-TOF mass spectrometry (multiphoton ionization, MALDI, ESI+), molecular beam high vacuum apparatus, high voltage electronics, molecular computational modeling with Gaussian, data acquisition with LabView, and data manipulation with Mathematica and Origin RESEARCH EXPERIENCE Graduate Student Researcher 2012-2017 • Time dependent (transient) photo relaxation of organic molecules, including PAHs and aromatic biological molecules.