GABA Receptors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Kava (Piper Methysticum) and Its Methysticin Constituents Protect Brain Tissue Against Ischemic Damage in Rodents
5 Refs: Arletti R et al, Stimulating property of Turnera diffusa and Pfaffia paniculata extracts on the sexual-behavior of male rats. Psychopharmacology 143(1), 15-19, 1999. Berger F, Handbuch der drogenkunde . Vol 2, Maudrich, Wien, 1950. Martinez M, Les plantas medicinales de Mexico . Cuarta Edicion Botas Mexico , p119, 1959. Tyler VE et al, Pharmacognosy , 9 th edition, Lea & Febiger, Philadelphia, 1988. KAVA ( Piper methysticum ) - A REVIEW The Kava plant (Piper methysticum) is a robust, well-branching and erect perennial shrub belonging to the pepper family (Piperaceae). The botanical origin remains unknown, although it is likely that early Polynesian explorers brought the plant with them from island to island. Numerous varieties of Kava exist, and today it is widely cultivated in several Pacific Island countries both for local use as well as the rapidly growing demand for pharmaceutical preparations. The dried rhizomes (roots) are normally used. The first description to the western world of the ceremonial use of an intoxicating beverage prepared from Kava was made by Captain James Cook following his Pacific voyage in 1768. The drink, prepared as an infusion in an elaborate manner after first chewing the root, is consumed on formal occasions or meetings of village elders and chiefs, as well as in reconciling with enemies and on a more social basis. It remains an important social custom in many Pacific Island countries today. Most of the islands of the Pacific possessed Kava prior to European contact, particularly those encompassed by Polynesia, Melanesia and Micronesia. After drinking the Kava beverage a pleasantly relaxed and sociable state develops, after which a deep and restful sleep occurs. -

Alternative Treatments for Depression and Anxiety
2019 PCB Conference: Strickland Benzodiazepines (BZDs), Herbal and Alternative Treatments for Anxiety & Depression BZD Learning Objectives • List at least three uses for benzodiazepines • Discuss at least two risk factors associated with benzodiazepine prescriptions Craig Strickland, PhD, Owner Biobehavioral Education and Consultation https://sites.google.com/site/bioedcon 1 2 BZD Pharmacokinetics Clinical Uses of BZDs Generic Name Trade Name Rapidity ½ Life Dose (mg) • Treat a variety of anxiety disorders alprazolam Xanax Intermediate Short 0.75-4 • Hypnotics • Muscle relaxants chlordiaze- Librium Intermediate Long 15-100 poxide • To produce anterograde amnesia clonazepam Klonopin Intermediate Long 0.5-4 • Alcohol & other CNS depressant withdrawal • Anti-convulsant therapy diazepam Valium Rapid Long 4-40 triazolam Halcion Intermediate Very short 0.125-0.5 temazepam Restoril Short Short 7.5-30 3 4 1 2019 PCB Conference: Strickland Issues with BZDs Herbal Medication and Alternative Therapies Used in the Treatment of Depression and Anxiety • Addictive potential • Confusion between “anti-anxiety” effects and the “warm-fuzzy) • Large dose ranges • Comparison of BZDs with medications like Buspar, etc. • They work, they work well and they work quickly 5 6 Alternative Tx. Learning Objectives Background Information on herbals: Natural does not necessarily mean “safe” • List several amino acid treatments for depression • Side-effects and adverse reactions • List at least three of the most common herbal – Herbal medications are “drugs” although -

Could Mycolactone Inspire New Potent Analgesics? Perspectives and Pitfalls
toxins Review Could Mycolactone Inspire New Potent Analgesics? Perspectives and Pitfalls 1 2 3, 4, , Marie-Line Reynaert , Denis Dupoiron , Edouard Yeramian y, Laurent Marsollier * y and 1, , Priscille Brodin * y 1 France Univ. Lille, CNRS, Inserm, CHU Lille, Institut Pasteur de Lille, U1019-UMR8204-CIIL-Center for Infection and Immunity of Lille, F-59000 Lille, France 2 Institut de Cancérologie de l’Ouest Paul Papin, 15 rue André Boquel-49055 Angers, France 3 Unité de Microbiologie Structurale, Institut Pasteur, CNRS, Univ. Paris, F-75015 Paris, France 4 Equipe ATIP AVENIR, CRCINA, INSERM, Univ. Nantes, Univ. Angers, 4 rue Larrey, F-49933 Angers, France * Correspondence: [email protected] (L.M.); [email protected] (P.B.) These three authors contribute equally to this work. y Received: 29 June 2019; Accepted: 3 September 2019; Published: 4 September 2019 Abstract: Pain currently represents the most common symptom for which medical attention is sought by patients. The available treatments have limited effectiveness and significant side-effects. In addition, most often, the duration of analgesia is short. Today, the handling of pain remains a major challenge. One promising alternative for the discovery of novel potent analgesics is to take inspiration from Mother Nature; in this context, the detailed investigation of the intriguing analgesia implemented in Buruli ulcer, an infectious disease caused by the bacterium Mycobacterium ulcerans and characterized by painless ulcerative lesions, seems particularly promising. More precisely, in this disease, the painless skin ulcers are caused by mycolactone, a polyketide lactone exotoxin. In fact, mycolactone exerts a wide range of effects on the host, besides being responsible for analgesia, as it has been shown notably to modulate the immune response or to provoke apoptosis. -
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
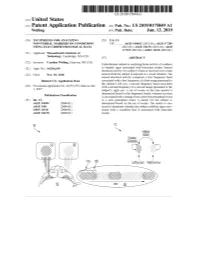
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

Benzodiazepines: Uses and Risks Charlie Reznikoff, MD Hennepin Healthcare
Benzodiazepines: Uses and Risks Charlie Reznikoff, MD Hennepin healthcare 4/22/2020 Overview benzodiazepines • Examples of benzos and benzo like drugs • Indications for benzos • Pharmacology of benzos • Side effects and contraindications • Benzo withdrawal • Benzo tapers 12/06/2018 Sedative/Hypnotics • Benzodiazepines • Alcohol • Z-drugs (Benzo-like sleeping aids) • Barbiturates • GHB • Propofol • Some inhalants • Gabapentin? Pregabalin? 12/06/2018 Examples of benzodiazepines • Midazolam (Versed) • Triazolam (Halcion) • Alprazolam (Xanax) • Lorazepam (Ativan) • Temazepam (Restoril) • Oxazepam (Serax) • Clonazepam (Klonopin) • Diazepam (Valium) • Chlordiazepoxide (Librium) 4/22/2020 Sedatives: gaba stimulating drugs have incomplete “cross tolerance” 12/06/2018 Effects from sedative (Benzo) use • Euphoria/bliss • Suppresses seizures • Amnesia • Muscle relaxation • Clumsiness, visio-spatial impairment • Sleep inducing • Respiratory suppression • Anxiolysis/disinhibition 12/06/2018 Tolerance to benzo effects? • Effects quickly diminish with repeated use (weeks) • Euphoria/bliss • Suppresses seizures • Effects incompletely diminish with repeated use • Amnesia • Muscle relaxation • Clumsiness, visio-spatial impairment • Seep inducing • Durable effects with repeated use • Respiratory suppression • Anxiolysis/disinhibition 12/06/2018 If you understand this pharmacology you can figure out the rest... • Potency • 1 mg diazepam <<< 1 mg alprazolam • Duration of action • Half life differences • Onset of action • Euphoria, clinical utility in acute -

Effect of Repeated Gaboxadol Administration on Night Sleep and Next-Day Performance in Healthy Elderly Subjects
Neuropsychopharmacology (2005) 30, 833–841 & 2005 Nature Publishing Group All rights reserved 0893-133X/05 $30.00 www.neuropsychopharmacology.org Effect of Repeated Gaboxadol Administration on Night Sleep and Next-Day Performance in Healthy Elderly Subjects 1 2 ,3 1 Stefan Mathias , Josef Zihl , Axel Steiger* and Marike Lancel 1Section of Sleep Pharmacology, Max-Planck-Institute of Psychiatry, Munich, Germany; 2Section of Neuropsychology, Max-Planck-Institute of Psychiatry, Munich, Germany; 3Department of Psychiatry, Max-Planck-Institute of Psychiatry, Munich, Germany Aging is associated with dramatic reductions in sleep continuity and sleep intensity. Since gaboxadol, a selective GABAA receptor agonist, has been demonstrated to improve sleep consolidation and promote deep sleep, it may be an effective hypnotic, particularly for elderly patients with insomnia. In the present study, we investigated the effects of subchronic gaboxadol administration on nocturnal sleep and its residual effects during the next days in elderly subjects. This was a randomized, double-blind, placebo-controlled, balanced crossover study in 10 healthy elderly subjects without sleep complaints. The subjects were administered either placebo or 15 mg gaboxadol hydrochloride at bedtime on three consecutive nights. Sleep was recorded during each night from 2300 to 0700 h and tests assessing attention (target detection, stroop test) and memory function (visual form recognition, immediate word recall, digit span) were applied at 0900, 1400, and 1700 h during the following days. Compared with placebo, gaboxadol significantly shortened subjective sleep onset latency and increased self-rated sleep intensity and quality. Polysomnographic recordings showed that it significantly decreased the number of awakenings, the amount of intermittent wakefulness, and stage 1, and increased slow wave sleep and stage 2. -

NIH Public Access Author Manuscript Synapse
NIH Public Access Author Manuscript Synapse. Author manuscript; available in PMC 2010 May 4. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Synapse. 2006 November ; 60(6): 411±419. doi:10.1002/syn.20314. GABAA Receptor Regulation of Voluntary Ethanol Drinking Requires PKCε Joyce Besheer, Veronique Lepoutre, Beth Mole, and Clyde W. Hodge* Bowles Center for Alcohol Studies, Department of Psychiatry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599 Abstract Protein kinase C (PKC) regulates a variety of neural functions, including ion channel activity, neurotransmitter release, receptor desensitization and differentiation. We have shown previously that mice lacking the ε-isoform of PKC (PKCε) self-administer 75% less ethanol and exhibit supersensitivity to acute ethanol and allosteric positive modulators of GABAA receptors when compared with wild-type controls. The purpose of the present study was to examine involvement of PKCε in GABAA receptor regulation of voluntary ethanol drinking. To address this question, PKCε null-mutant and wild-type control mice were allowed to drink ethanol (10% v/v) vs. water on a two-bottle continuous access protocol. The effects of diazepam (nonselective GABAA BZ positive modulator), zolpidem (GABAA α1 agonist), L-655,708 (BZ-sensitive GABAA α5 inverse agonist), and flumazenil (BZ antagonist) were then tested on ethanol drinking. Ethanol intake (grams/kg/day) by wild-type mice decreased significantly after diazepam or zolpidem but increased after L-655,708 administration. Flumazenil antagonized diazepam-induced reductions in ethanol drinking in wild- type mice. However, ethanol intake by PKCε null mice was not altered by any of the GABAergic compounds even though effects were seen on water drinking in these mice. -

Valerenic Acid Potentiates and Inhibits GABAA Receptors: Molecular Mechanism and Subunit Specificity
ARTICLE IN PRESS + MODEL Neuropharmacology xx (2007) 1e10 www.elsevier.com/locate/neuropharm Valerenic acid potentiates and inhibits GABAA receptors: Molecular mechanism and subunit specificity S. Khom a, I. Baburin a, E. Timin a, A. Hohaus a, G. Trauner b, B. Kopp b, S. Hering a,* a Department of Pharmacology and Toxicology, University of Vienna, Althanstrasse 14, A-1090 Vienna, Austria b Department of Pharmacognosy, University of Vienna, Althanstrasse 14, A-1090 Vienna, Austria Received 8 December 2006; received in revised form 11 April 2007; accepted 30 April 2007 Abstract Valerian is a commonly used herbal medicinal product for the treatment of anxiety and insomnia. Here we report the stimulation of chloride currents through GABAA receptors (IGABA) by valerenic acid (VA), a constituent of Valerian. To analyse the molecular basis of VA action, we expressed GABAA receptors with 13 different subunit compositions in Xenopus oocytes and measured IGABA using the two-microelectrode voltage-clamp technique. We report a subtype-dependent stimulation of IGABA by VA. Only channels incorporating b2 or b3 subunits were stimulated by VA. Replacing b2/3 by b1 drastically reduced the sensitivity of the resulting GABAA channels. The stimulatory effect of VA on a1b2 receptors was substantially reduced by the point mutation b2N265S (known to inhibit loreclezole action). Mutating the corresponding residue of b1 (b1S290N) induced VA sensitivity in a1b1S290N comparable to a1b2 receptors. Modulation of IGABA was not significantly dependent on incorporation of a1, a2, a3 or a5 subunits. VA displayed a significantly lower efficiency on channels incorporating a4 subunits. IGABA modulation by VA was not g subunit dependent and not inhibited by flumazenil (1 mM). -

Molecular Dissection of G-Protein Coupled Receptor Signaling and Oligomerization
MOLECULAR DISSECTION OF G-PROTEIN COUPLED RECEPTOR SIGNALING AND OLIGOMERIZATION BY MICHAEL RIZZO A Dissertation Submitted to the Graduate Faculty of WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Biology December, 2019 Winston-Salem, North Carolina Approved By: Erik C. Johnson, Ph.D. Advisor Wayne E. Pratt, Ph.D. Chair Pat C. Lord, Ph.D. Gloria K. Muday, Ph.D. Ke Zhang, Ph.D. ACKNOWLEDGEMENTS I would first like to thank my advisor, Dr. Erik Johnson, for his support, expertise, and leadership during my time in his lab. Without him, the work herein would not be possible. I would also like to thank the members of my committee, Dr. Gloria Muday, Dr. Ke Zhang, Dr. Wayne Pratt, and Dr. Pat Lord, for their guidance and advice that helped improve the quality of the research presented here. I would also like to thank members of the Johnson lab, both past and present, for being valuable colleagues and friends. I would especially like to thank Dr. Jason Braco, Dr. Jon Fisher, Dr. Jake Saunders, and Becky Perry, all of whom spent a great deal of time offering me advice, proofreading grants and manuscripts, and overall supporting me through the ups and downs of the research process. Finally, I would like to thank my family, both for instilling in me a passion for knowledge and education, and for their continued support. In particular, I would like to thank my wife Emerald – I am forever indebted to you for your support throughout this process, and I will never forget the sacrifices you made to help me get to where I am today. -

GABA Receptors
D Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews Review No.7 / 1-2011 GABA receptors Wolfgang Froestl , CNS & Chemistry Expert, AC Immune SA, PSE Building B - EPFL, CH-1015 Lausanne, Phone: +41 21 693 91 43, FAX: +41 21 693 91 20, E-mail: [email protected] GABA Activation of the GABA A receptor leads to an influx of chloride GABA ( -aminobutyric acid; Figure 1) is the most important and ions and to a hyperpolarization of the membrane. 16 subunits with γ most abundant inhibitory neurotransmitter in the mammalian molecular weights between 50 and 65 kD have been identified brain 1,2 , where it was first discovered in 1950 3-5 . It is a small achiral so far, 6 subunits, 3 subunits, 3 subunits, and the , , α β γ δ ε θ molecule with molecular weight of 103 g/mol and high water solu - and subunits 8,9 . π bility. At 25°C one gram of water can dissolve 1.3 grams of GABA. 2 Such a hydrophilic molecule (log P = -2.13, PSA = 63.3 Å ) cannot In the meantime all GABA A receptor binding sites have been eluci - cross the blood brain barrier. It is produced in the brain by decarb- dated in great detail. The GABA site is located at the interface oxylation of L-glutamic acid by the enzyme glutamic acid decarb- between and subunits. Benzodiazepines interact with subunit α β oxylase (GAD, EC 4.1.1.15). It is a neutral amino acid with pK = combinations ( ) ( ) , which is the most abundant combi - 1 α1 2 β2 2 γ2 4.23 and pK = 10.43. -

Benzodiazepine Actions Mediated by Specificg-Aminobutyric Acida
letters to nature ..................................................................................................................................... erratum Benzodiazepine actions mediated by speci®c g-aminobutyric acidA receptor subtypes Uwe Rudolph, Florence Crestani, Dietmar Benke, Ina BruÈnig, Jack A. Benson, Jean-Marc Fritschy, James R. Martin, Horst Bluethmann & Hanns MoÈhler Nature 401, 796±800 (1999) .......................................................................................................................................................................................................................................................................... The quality of Fig. 2 was unsatisfactory as published. The ®gure is reproduced again here. M 3 3 a d [ H]Ro15-4513 [ H]Ro15-4513, Ki (nM) + diazepam 1(H101R) 1(H101R) α α Wild-type Wild-type α1 α5 Wild-type α2 β2/3 α3 γ2 1(H101R) α α b Wild-type 1(H101R) e Wild-type 3 µM GABA+Dz +Ro15 3 µM GABA α1 α1(H101R) 3 µ µ c Displacement of [ H]Ro15-4513, Ki (nM) 3 M GABA+Dz +Ro15 3 M GABA Wild-type α1(H101R) Clonazepam 3 ± 2 > 10,000 Diazepam 28 ± 15 > 10,000 Zolpidem 13 ± 5 > 10,000 500 pA 2 s ................................................................. suggest that pairing occurs primarily on the electrons on the g- correction sheet of the Fermi surface under the conditions of the experiment. However, the orientation of the FLL, interpreted within Agterberg's two-component Ginzburg±Landau (TCGL) theory1,2, is no longer consistent with g-band pairing if we take the values of Fermi surface Observation of a square ¯ux-line anisotropy used in refs 1, 2, and obtained by recent band structure calculations3. However, other calculations (T. Oguchi, private lattice in the unconventional communication) give a different sign of Fermi surface anisotropy. TCGL theory predictions may be modi®ed by extra anisotropy of superconductor Sr2RuO4 the superconducting energy gap3, by substantial pairing on the a- and/or b-sheets4 or by anisotropy in the electron mass enhance- T. -

Assessment Report on Salvia Officinalis L., Folium and Salvia Officinalis L., Aetheroleum Final
20 September 2016 EMA/HMPC/150801/2015 Committee on Herbal Medicinal Products (HMPC) Assessment report on Salvia officinalis L., folium and Salvia officinalis L., aetheroleum Final Based on Article 16d(1), Article 16f and Article 16h of Directive 2001/83/EC (traditional use) Herbal substance(s) (binomial scientific name of Salvia officinalis L., folium and the plant, including plant part) Salvia officinalis L., aetheroleum Herbal preparation(s) a) Comminuted herbal substance b) Liquid extract (DER 1:1), extraction solvent ethanol 70% V/V c) Dry extract (DER 4-7:1), extraction solvent water d) Liquid extract (DER 1:3.5-5), extraction solvent ethanol 31.5% V/V e) Liquid extract (DER 1:4-5) extraction solvent ethanol 50% V/V f) Liquid extract (DER 1:4-6), extraction solvent liquor wine:ethanol 96% V/V (38.25:61.75 m/m) g) Tincture (ratio of herbal substance to extraction solvent 1:10) extraction solvent ethanol 70% V/V Pharmaceutical form(s) Comminuted herbal substance as herbal tea for oral use. Comminuted herbal substance for infusion preparation for oromucosal or cutaneous use. Herbal preparations in solid or liquid dosage forms for oral use. Herbal preparations in liquid or semi-solid dosage forms for cutaneous use or for oromucosal use. 30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5555 Send a question via our website www.ema.europa.eu/contact An agency of the European Union © European Medicines Agency, 2017. Reproduction is authorised provided the source is acknowledged.