Mossbauer Spectroscopy of 57Fe
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Comparing the Emission Spectra of U and Th Hollow Cathode Lamps and a New U Line-List
Astronomy & Astrophysics manuscript no. HCL_U_Th_Ull_S2018 c ESO 2018 October 1, 2018 Comparing the emission spectra of U and Th hollow cathode lamps and a new U line-list L. F. Sarmiento1,⋆, A. Reiners1, P. Huke1, F. F. Bauer1, E. W. Guenter2,3, U. Seemann1, and U. Wolter4 1 Institut für Astrophysik, Georg-August-Universität Göttingen, Friedrich-Hund-Platz 1, 37077 Göttingen, Germany 2 Thüringer Landessternwarte Tautenburg, Sternwarte 5, 07778 Tautenburg, Germany 3 Instituto de Astrofísica de Canarias (IAC), 38205 La Laguna, Tenerife, Spain 4 Hamburger Sternwarte, Universität Hamburg, Gojenbergsweg 112, 21029 Hamburg, Germany Received , 22 February 2018; accepted , 5 June 2018 ABSTRACT Context. Thorium hollow cathode lamps (HCLs) are used as frequency calibrators for many high resolution astronomical spectro- graphs, some of which aim for Doppler precision at the 1 m/s level. Aims. We aim to determine the most suitable combination of elements (Th or U, Ar or Ne) for wavelength calibration of astronomical spectrographs, to characterize differences between similar HCLs, and to provide a new U line-list. Methods. We record high resolution spectra of different HCLs using a Fourier transform spectrograph: (i) U-Ne, U-Ar, Th-Ne, and Th-Ar lamps in the spectral range from 500 to 1000 nm and U-Ne and U-Ar from 1000 to 1700 nm; (ii) we systematically compare the number of emission lines and the line intensity ratio for a set of 12 U-Ne HCLs; and (iii) we record a master spectrum of U-Ne to create a new U line-list. Results. Uranium lamps show more lines suitable for calibration than Th lamps from 500 to 1000 nm. -

Mössbauer Spectroscopy 9.1 Recoil Free Resonance Absorption
Chapter 9, page 1 9 Mössbauer Spectroscopy 9.1 Recoil free resonance absorption Robert Wood published in 1905 an article "Resonance Radiation of Sodium Vapor" (see Literature) and reported that if a bulb containing pure sodium vapor was illuminated by light from a sodium flame, the vapor emitted a yellow light which spectroscopic analysis showed to be identical with the exciting light, in other words, the two D lines. Sodium is liquid above 98 °C, boiling point 883 °C, thus a remarkable vapor pressure exists, if sodium is heated by the Fig. 9.1: Wood's apparatus for the Bunsen burner above 200 °C. Wood used the name resonance radiation of the sodium "resonance radiation" for the effect of identical D lines. absorbed radiation and fluorescence radiation. Now we use the term "resonance absorption" instead. Since 1900 γ-rays were known as a highly energetic monochromatic radiation, but the anticipated γ-ray resonance fluorescence failed to occur. Werner Kuhn succeeded in publishing such an experiment that don't work and argued in 1929: "The (third) influence, reducing the absorption, arises from the emission process of the γ-rays. The emitting atom will suffer recoil due to the projection of the γ-ray. The wavelength of the radiation is therefore shifted to the red; the emission line is displaced relative to the absorption line.... It is thus possible that by a large γ-shift, the whole emission line is brought out of the absorption region”. (see Literature, Kuhn) Intensity In the case of γ-radiation the atoms suffer recoil, which is not significant for radiation in the visible range, were the recoil energy is small compared with the linewidth of the h δν1/2 radiation (times h), see Fig. -

Stark Broadening of Spectral Lines in Plasmas
atoms Stark Broadening of Spectral Lines in Plasmas Edited by Eugene Oks Printed Edition of the Special Issue Published in Atoms www.mdpi.com/journal/atoms Stark Broadening of Spectral Lines in Plasmas Stark Broadening of Spectral Lines in Plasmas Special Issue Editor Eugene Oks MDPI • Basel • Beijing • Wuhan • Barcelona • Belgrade Special Issue Editor Eugene Oks Auburn University USA Editorial Office MDPI St. Alban-Anlage 66 4052 Basel, Switzerland This is a reprint of articles from the Special Issue published online in the open access journal Atoms (ISSN 2218-2004) in 2018 (available at: https://www.mdpi.com/journal/atoms/special issues/ stark broadening plasmas) For citation purposes, cite each article independently as indicated on the article page online and as indicated below: LastName, A.A.; LastName, B.B.; LastName, C.C. Article Title. Journal Name Year, Article Number, Page Range. ISBN 978-3-03897-455-0 (Pbk) ISBN 978-3-03897-456-7 (PDF) Cover image courtesy of Eugene Oks. c 2018 by the authors. Articles in this book are Open Access and distributed under the Creative Commons Attribution (CC BY) license, which allows users to download, copy and build upon published articles, as long as the author and publisher are properly credited, which ensures maximum dissemination and a wider impact of our publications. The book as a whole is distributed by MDPI under the terms and conditions of the Creative Commons license CC BY-NC-ND. Contents About the Special Issue Editor ...................................... vii Preface to ”Stark Broadening of Spectral Lines in Plasmas” ..................... ix Eugene Oks Review of Recent Advances in the Analytical Theory of Stark Broadening of Hydrogenic Spectral Lines in Plasmas: Applications to Laboratory Discharges and Astrophysical Objects Reprinted from: Atoms 2018, 6, 50, doi:10.3390/atoms6030050 ................... -

Quantitative Analysis of Cerium-Gallium Alloys Using a Hand-Held Laser Induced Breakdown Spectroscopy Device
atoms Article Quantitative Analysis of Cerium-Gallium Alloys Using a Hand-Held Laser Induced Breakdown Spectroscopy Device Ashwin P. Rao 1 , Matthew T. Cook 2 , Howard L. Hall 2,3 and Michael B. Shattan 1,* 1 Air Force Institute of Technology, 2950 Hobson Way, WPAFB, OH 45433, USA 2 Institute for Nuclear Security, University of Tennessee, Knoxville, TN 37996, USA 3 Department of Nuclear Engineering, University of Tennessee, Knoxville, TN 37996, USA * Correspondence: michael.shattan@afit.edu Received: 26 July 2019; Accepted: 20 August 2019; Published: 22 August 2019 Abstract: A hand-held laser-induced breakdown spectroscopy device was used to acquire spectral emission data from laser-induced plasmas created on the surface of cerium-gallium alloy samples with Ga concentrations ranging from 0–3 weight percent. Ionic and neutral emission lines of the two constituent elements were then extracted and used to generate calibration curves relating the emission line intensity ratios to the gallium concentration of the alloy. The Ga I 287.4-nm emission line was determined to be superior for the purposes of Ga detection and concentration determination. A limit of detection below 0.25% was achieved using a multivariate regression model of the Ga I 287.4-nm line ratio versus two separate Ce II emission lines. This LOD is considered a conservative estimation of the technique’s capability given the type of the calibration samples available and the low power (5 mJ per 1-ns pulse) and resolving power (l/Dl = 4000) of this hand-held device. Nonetheless, the utility of the technique is demonstrated via a detailed mapping analysis of the surface Ga distribution of a Ce-Ga sample, which reveals significant heterogeneity resulting from the sample production process. -

Emission Mössbauer Spectroscopy at ISOLDE/CERN
Emission Mössbauer Spectroscopy at ISOLDE/CERN Torben Esmann Mølholt ISOLDE Seminar, 25. Nov. 2015 Outline Experimental setup at ISOLDE Brief on the Mössbauer spectroscopy technique Examples and Results Future/ongoing measurements 2 Acknowledgements The Mössbauer collaboration at ISOLDE/CERN, >30 active members with new members (2014) from China, Russia, Bulgaria, Austria, Spain: Four experiments Existing members New members 2014 (IS-501, IS-576, IS-578, I-161) 3 Emission Mössbauer Spectroscopy at ISOLDE/CERN http://e-ms.web.cern.ch/ GLM (GPS) LA1-2 (HRS) 4 119In RILIS 2014 2015 57Mn 119 RILIS In 15 μSi/h - 57Mn 10 μSi/h - RILIS 5 μSi/h - 0 μSi/h - 5 Mössbauer Experimental setup Implantation chamber Incoming 60 keV beam Sample Faraday cup Be window Mössbauer drive with resonance detector Container: 25 mbar acetone • Intensity (~1×108 atoms/s) • High statistics spectrum (5 – 10 min.) •On-line (short lived) •Collections for Off-line (long lived) 6 •Hours - days Mössbauer Experimental setup Sample holder •Temperature range 90 – 700 K • Measurements at different emission angles • Applied magnetic field (Bext ≤ 0.6 T) 7 Mössbauer Experimental setup Sample holder • Quenching: Implant at high temperature Measure at low temperature (off-line) 8 Mössbauer Experimental setup Resonance detector - G. Weyer, Mössbauer Eff. Meth., 10 (1976) 301 PPAD: Parallel Plate Avalanche Detector - Single line resonance detector. 0.1 cps (~0.1 µCi) – 50k cps (~500 mCi) 9 Mössbauer spectroscopy technique 10 40-60 keV Ion-implantation of Mössbauer Probe Emission -

Glossary of Terms Absorption Line a Dark Line at a Particular Wavelength Superimposed Upon a Bright, Continuous Spectrum
Glossary of terms absorption line A dark line at a particular wavelength superimposed upon a bright, continuous spectrum. Such a spectral line can be formed when electromag- netic radiation, while travelling on its way to an observer, meets a substance; if that substance can absorb energy at that particular wavelength then the observer sees an absorption line. Compare with emission line. accretion disk A disk of gas or dust orbiting a massive object such as a star, a stellar-mass black hole or an active galactic nucleus. An accretion disk plays an important role in the formation of a planetary system around a young star. An accretion disk around a supermassive black hole is thought to be the key mecha- nism powering an active galactic nucleus. active galactic nucleus (agn) A compact region at the center of a galaxy that emits vast amounts of electromagnetic radiation and fast-moving jets of particles; an agn can outshine the rest of the galaxy despite being hardly larger in volume than the Solar System. Various classes of agn exist, including quasars and Seyfert galaxies, but in each case the energy is believed to be generated as matter accretes onto a supermassive black hole. adaptive optics A technique used by large ground-based optical telescopes to remove the blurring affects caused by Earth’s atmosphere. Light from a guide star is used as a calibration source; a complicated system of software and hardware then deforms a small mirror to correct for atmospheric distortions. The mirror shape changes more quickly than the atmosphere itself fluctuates. -

Arxiv:Cond-Mat/0404626V1
LA-UR-03-8735 Nuclear Magnetic Resonance Studies of δ-Stabilized Plutonium N. J. Curro Condensed Matter and Thermal Physics, Los Alamos National Laboratory, Los Alamos, NM 87545, [email protected] L. Morales Nuclear Materials and Technology Division, Los Alamos National Laboratory, Los Alamos, NM 87545 (Dated: July 14, 2018) Nuclear Magnetic Resonance studies of Ga stabilized δ-Pu reveal detailed information about the local distortions surrounding the Ga impurities as well as provides information about the local spin fluctuations experienced by the Ga nuclei. The Ga NMR spectrum is inhomogeneously broadened by a distribution of local electric field gradients (EFGs), which indicates that the Ga experiences local distortions from cubic symmetry. The Knight shift and spin lattice relaxation rate indicate that the Ga is dominantly coupled to the Fermi surface via core polarization, and is inconsistent with magnetic order or low frequency spin correlations. Introduction tuations of of the electron spin S relax the nuclei so fast that their signal is rendered invisible. On the other hand, The investigavtion of the low temperature properties the hyperfine coupling to the nuclei of the secondary el- of plutonium and its compounds has experienced a re- ement (Al, Ga or In) are typically one to three orders of naissance in recent years, and several important experi- magnitude smaller, so by measuring the secondary nuclei ments have revealed unusual correlated electron behavior one can gain considerable insight into the spin dynamics [1, 2, 3]. The 5f electrons in elemental plutonium are on of the system. the boundary between localized and itinerant behavior, One of the challenges facing band theorists calculating so that slight perturbations in the Pu-Pu spacing can give the electronic structure of δ-Pu is the role of the sec- rise to dramatic changes in the ground state character. -

Rapid Analysis of Plutonium Surrogate Material Via Hand-Held Laser-Induced Breakdown Spectroscopy
Air Force Institute of Technology AFIT Scholar Theses and Dissertations Student Graduate Works 3-2020 Rapid Analysis of Plutonium Surrogate Material via Hand-Held Laser-Induced Breakdown Spectroscopy Ashwin P. Rao Follow this and additional works at: https://scholar.afit.edu/etd Part of the Atomic, Molecular and Optical Physics Commons, and the Nuclear Engineering Commons Recommended Citation Rao, Ashwin P., "Rapid Analysis of Plutonium Surrogate Material via Hand-Held Laser-Induced Breakdown Spectroscopy" (2020). Theses and Dissertations. 3599. https://scholar.afit.edu/etd/3599 This Thesis is brought to you for free and open access by the Student Graduate Works at AFIT Scholar. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of AFIT Scholar. For more information, please contact [email protected]. RAPID ANALYSIS OF PLUTONIUM SURROGATE MATERIAL VIA HAND-HELD LASER-INDUCED BREAKDOWN SPECTROSCOPY THESIS Ashwin P. Rao, Second Lieutenant, USAF AFIT-ENP-MS-20-M-115 DEPARTMENT OF THE AIR FORCE AIR UNIVERSITY AIR FORCE INSTITUTE OF TECHNOLOGY Wright-Patterson Air Force Base, Ohio DISTRIBUTION STATEMENT A APPROVED FOR PUBLIC RELEASE; DISTRIBUTION UNLIMITED. The views expressed in this thesis are those of the author and do not reflect the official policy or position of the United States Air Force, Department of Defense, or the United States Government. This material is declared a work of the U.S. Government and is not subject to copyright protection in the United States. AFIT-ENP-MS-20-M-115 RAPID ANALYSIS OF PLUTONIUM SURROGATE MATERIAL VIA HAND-HELD LASER-INDUCED BREAKDOWN SPECTROSCOPY THESIS Presented to the Faculty Department of Engineering Physics Graduate School of Engineering and Management Air Force Institute of Technology Air University Air Education and Training Command in Partial Fulfillment of the Requirements for the Degree of Master of Science in Nuclear Engineering Ashwin P. -

Ground-State Wave Function of Plutonium in Pusb As Determined Via X-Ray Magnetic Circular Dichroism
PHYSICAL REVIEW B 91, 035117 (2015) Ground-state wave function of plutonium in PuSb as determined via x-ray magnetic circular dichroism M. Janoschek,1,* D. Haskel,2 J. Fernandez-Rodriguez,2,3 M. van Veenendaal,2,3 J. Rebizant,4 G. H. Lander,4 J.-X. Zhu,1 J. D. Thompson,1 and E. D. Bauer1 1Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA 2Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois 60439, USA 3Department of Physics, Northern Illinois University, DeKalb, Illinois 60115, USA 4European Commission, JRC, Institute for Transuranium Elements, 76125 Karlsruhe, Germany (Received 27 October 2014; published 14 January 2015) Measurements of x-ray magnetic circular dichroism (XMCD) and x-ray absorption near-edge structure (XANES) spectroscopy at the Pu M4,5 edges of the ferromagnet PuSb are reported. Using bulk magnetization measurements and a sum rule analysis of the XMCD spectra, we determine the individual orbital [μL = 2.8(1)μB/Pu] and spin moments [μS =−2.0(1)μB/Pu] of the Pu 5f electrons. Atomic multiplet calculations of the XMCD and XANES spectra reproduce well the experimental data and are consistent with the experimental value of the spin moment. These measurements of Lz and Sz are in excellent agreement with the values that have been extracted from neutron magnetic form factor measurements, and confirm the local character of the 5f electrons in PuSb. Finally, we demonstrate that a split M5 as well as a narrow M4 XMCD signal may serve as a signature of 5f electron localization in actinide compounds. DOI: 10.1103/PhysRevB.91.035117 PACS number(s): 75.25.−j, 78.70.Dm, 71.20.Lp, 71.27.+a I. -

Antisite Defects in Epitaxial Films of Lutetium Doped Yttrium Iron Garnets Studied by Nuclear Magnetic Resonance V
Vol. 118 (2010) ACTA PHYSICA POLONICA A No. 5 14th Czech and Slovak Conference on Magnetism, Košice, Slovakia, July 6–9, 2010 Antisite Defects in Epitaxial Films of Lutetium Doped Yttrium Iron Garnets Studied by Nuclear Magnetic Resonance V. Chlana, H. Štěpánkováa, J. Englicha, R. Řezníčeka, K. Kouřila, M. Kučeraa and K. Nitschb aCharles University in Prague, Faculty of Mathematics and Physics V Holešovičkách 2, 18000, Prague 8, Czech Republic bInstitute of Physics, ASCR, Cukrovarnická 10, 162 53 Prague 6, Czech Republic Series of lutetium doped yttrium iron garnet films is studied by means of 57Fe nuclear magnetic resonance. Satellite spectral lines are resolved and identified in the spectra and concentrations of lutetium in dodecahedral sites as well as yttrium/lutetium antisite defects in octahedral sites are estimated. Compared to yttrium, lutetium cations are found to have stronger disposition towards creating the antisite defects. PACS numbers: 75.50.Gg, 76.60.–k, 76.60.Lz, 68.35.Dv, 68.55.Ln 1. Introduction used. 57Fe NMR frequency-swept spectra of lutetium doped YIG garnets were measured at 4.2 K in zero mag- Garnet films can be designed for various applications netic field; spectral region corresponding to resonance of in optical, magnetooptical, and microwave devices [1, 2], irons in tetrahedral d-sites is displayed in Fig. 1. where one of the important factors concerns information on intrinsic defects and substitutions [3, 4]. Such knowl- edge may be well acquired by nuclear magnetic resonance (NMR) spectroscopy for its high sensitivity to local struc- ture perturbations. The NMR method is particularly suitable for detection of intrinsic defects, impurities or aimed substitutions. -

Electrical Engineering Dictionary
ratio of the power per unit solid angle scat- tered in a specific direction of the power unit area in a plane wave incident on the scatterer R from a specified direction. RADHAZ radiation hazards to personnel as defined in ANSI/C95.1-1991 IEEE Stan- RS commonly used symbol for source dard Safety Levels with Respect to Human impedance. Exposure to Radio Frequency Electromag- netic Fields, 3 kHz to 300 GHz. RT commonly used symbol for transfor- mation ratio. radial basis function network a fully R-ALOHA See reservation ALOHA. connected feedforward network with a sin- gle hidden layer of neurons each of which RL Typical symbol for load resistance. computes a nonlinear decreasing function of the distance between its received input and Rabi frequency the characteristic cou- a “center point.” This function is generally pling strength between a near-resonant elec- bell-shaped and has a different center point tromagnetic field and two states of a quan- for each neuron. The center points and the tum mechanical system. For example, the widths of the bell shapes are learned from Rabi frequency of an electric dipole allowed training data. The input weights usually have transition is equal to µE/hbar, where µ is the fixed values and may be prescribed on the electric dipole moment and E is the maxi- basis of prior knowledge. The outputs have mum electric field amplitude. In a strongly linear characteristics, and their weights are driven 2-level system, the Rabi frequency is computed during training. equal to the rate at which population oscil- lates between the ground and excited states. -
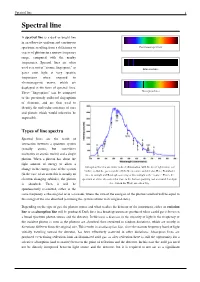
Spectral Line-Wikipedia.Pdf
Spectral line 1 Spectral line A spectral line is a dark or bright line in an otherwise uniform and continuous spectrum, resulting from a deficiency or Continuous spectrum excess of photons in a narrow frequency range, compared with the nearby frequencies. Spectral lines are often used as a sort of "atomic fingerprint," as Emission lines gases emit light at very specific frequencies when exposed to electromagnetic waves, which are displayed in the form of spectral lines. These "fingerprints" can be compared Absorption lines to the previously collected fingerprints of elements, and are thus used to identify the molecular construct of stars and planets which would otherwise be impossible. Types of line spectra Spectral lines are the result of interaction between a quantum system (usually atoms, but sometimes molecules or atomic nuclei) and a single photon. When a photon has about the right amount of energy to allow a Absorption lines for air, under indirect illumination, with the direct light source not change in the energy state of the system visible, so that the gas is not directly between source and detector. Here, Fraunhofer (in the case of an atom this is usually an lines in sunlight and Rayleigh scattering of this sunlight is the "source." This is the electron changing orbitals), the photon spectrum of a blue sky somewhat close to the horizon, pointing east at around 3 or 4 pm is absorbed. Then it will be (i.e., Sun in the West) on a clear day. spontaneously re-emitted, either in the same frequency as the original or in a cascade, where the sum of the energies of the photons emitted will be equal to the energy of the one absorbed (assuming the system returns to its original state).