Can the Olive Genome Be a Miracle for Human DNA?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tsrna Signatures in Cancer
tsRNA signatures in cancer Veronica Balattia, Giovanni Nigitaa,1, Dario Venezianoa,1, Alessandra Druscoa, Gary S. Steinb,c, Terri L. Messierb,c, Nicholas H. Farinab,c, Jane B. Lianb,c, Luisa Tomaselloa, Chang-gong Liud, Alexey Palamarchuka, Jonathan R. Harte, Catherine Belle, Mariantonia Carosif, Edoardo Pescarmonaf, Letizia Perracchiof, Maria Diodorof, Andrea Russof, Anna Antenuccif, Paolo Viscaf, Antonio Ciardig, Curtis C. Harrish, Peter K. Vogte, Yuri Pekarskya,2, and Carlo M. Crocea,2 aDepartment of Cancer Biology and Medical Genetics, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; bDepartment of Biochemistry, University of Vermont College of Medicine, Burlington, VT 05405; cUniversity of Vermont Cancer Center, College of Medicine, Burlington, VT 05405; dMD Anderson Cancer Center, Houston, TX 77030; eDepartment of Molecular Medicine, The Scripps Research Institute, La Jolla, CA 92037; fIstituto di Ricovero e Cura a Carattere Scientifico, Regina Elena National Cancer Institute, 00144 Rome, Italy; gUniversita’ Di Roma La Sapienza, 00185 Rome, Italy; and hLaboratory of Human Carcinogenesis, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892 Contributed by Carlo M. Croce, June 13, 2017 (sent for review April 26, 2017; reviewed by Riccardo Dalla-Favera and Philip N. Tsichlis) Small, noncoding RNAs are short untranslated RNA molecules, some these molecules, which we defined as single-stranded small of which have been associated with cancer development. Recently RNAs, 16–48 nt long, ending with a stretch of four Ts (4). When we showed that a class of small RNAs generated during the matu- tsRNAs accumulate in the nucleus, they can be exported, sug- ration process of tRNAs (tRNA-derived small RNAs, hereafter gesting that tsRNAs could regulate gene expression at different “tsRNAs”) is dysregulated in cancer. -

2020.05.13.092460V1.Full.Pdf
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.13.092460; this version posted May 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. An adaptive pre-DNA-damage-response protects genome integrity Sandrine Ragu1,2, Gabriel Matos-Rodrigues1,2, Nathalie Droin3, Aurélia Barascu1, Sylvain Caillat4, Gabriella Zarkovic5, Capucine Siberchicot6, Elodie Dardillac1,2, Camille Gelot1, Juan Pablo Radicella6, Alexander A. Ishchenko5, Jean-Luc Ravanat4, Eric Solary3 and Bernard S. Lopez1,2 * 1Institut Cochin, INSERM U1016, UMR 8104 CNRS, Université de Paris, Paris-Descartes, Equipe Labellisée Ligue Contre le Cancer 24 rue du Faubourg St Jacques, 75014 Paris, France 2CNRS UMR 8200, Institut de Cancérologie Gustave-Roussy, Université Paris Saclay, 114 Rue Edouard Vaillant, 94805 Villejuif, France. 3Institut de Cancérologie Gustave-Roussy, INSERM U1170, Université Paris Saclay, Equipe Labellisée Ligue Contre le Cancer 114 Rue Edouard Vaillant, 94805 Villejuif, France. 4CEA, INAC-SCIB-LAN, Université Grenoble Alpes, F-38000 Grenoble. 5 Groupe «Réparation de l'ADN», Equipe Labellisée par la Ligue Nationale contre le Cancer, CNRS UMR 8200, Gustave Roussy, Université Paris Saclay, F-94805 Villejuif, France 6Institute of Cellular and Molecular Radiobiology, Inst. de Biologie François Jacob, CEA, Université de Paris, Université Paris-Saclay, F-92260 Fontenay aux Roses, France *Correspondence to: Bernard S. Lopez. [email protected] Running title: Adapt cell response to stress severity 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.05.13.092460; this version posted May 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Histone H3.1 (Human) Cell-Based ELISA Kit
Histone H3.1 (Human) Cell-Based ELISA Kit Catalog # : KA2761 規格 : [ 1 Kit ] List All Specification Application Image Product Histone H3.1 (Human) Cell-Based ELISA Kit is an indirect enzyme-linked Qualitative Description: immunoassay for qualitative determination of Histone H3 expression in cultured cells. Reactivity: Human, Mouse, Rat Storage Store the kit at 4°C. Instruction: Protocol: Protocol Download Suitable Attached Cell, Loosely Attached Cell, Suspension Cell Sample: Label: HRP-conjugated Detection Colorimetric Method: Regulation For research use only (RUO) Status: Datasheet: Download Applications Qualitative HIST1H3A HIST1H3D HIST1H3C HIST1H3E HIST1H3I HIST1H3G HIST1H3J HIST1H3H HIST1H3B HIST1H3F Gene Information Entrez GeneID: 8350 Protein P68431 Accession#: Gene Name: HIST1H3A Gene Alias: H3/A,H3FA Gene histone cluster 1, H3a Description: Omim ID: 602810 Gene Ontology: Hyperlink Gene Summary: Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. This structure consists of approximately 146 bp of DNA wrapped around a Page 1 of 6 2021/6/18 nucleosome, an octamer composed of pairs of each of the four core histones (H2A, H2B, H3, and H4). The chromatin fiber is further compacted through the interaction of a linker histone, H1, with the DNA between the nucleosomes to form higher order chromatin structures. This gene is intronless and encodes a member of the histone H3 family. Transcripts from this gene lack polyA tails; instead, they contain a palindromic termination element. This gene is found in the large histone gene cluster on chromosome 6p22-p21.3. [provided by RefSeq Other H3 histone family, member A,histone 1, H3a Designations: Gene Information Entrez GeneID: 8351 Protein P68431 Accession#: Gene Name: HIST1H3D Gene Alias: H3/b,H3FB Gene histone cluster 1, H3d Description: Omim ID: 602811 Gene Ontology: Hyperlink Gene Summary: Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. -

Hidden Markov Models Predict Epigenetic Chromatin Domains
Hidden Markov Models Predict Epigenetic Chromatin Domains The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Larson, Jessica. 2012. Hidden Markov Models Predict Epigenetic Chromatin Domains. Doctoral dissertation, Harvard University. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:10087396 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ©2012- Jessica Lynn Larson All rights reserved Dissertation Advisor: Professor Guo-Cheng Yuan Jessica Lynn Larson Hidden Markov Models Predict Epigenetic Chromatin Domains Abstract Epigenetics is an important layer of transcriptional control necessary for cell-type specific gene regulation. We developed computational methods to analyze the combinatorial effect and large-scale organizations of genome-wide distributions of epigenetic marks. Throughout this dissertation, we show that regions containing multiple genes with similar epigenetic patterns are found throughout the genome, suggesting the presence of several chromatin domains. In Chapter 1, we develop a hidden Markov model (HMM) for detecting the types and locations of epigenetic domains from multiple histone modifications. We use this method to analyze a published ChIP-seq dataset of five histone modification marks in mouse embryonic stem cells. We successfully detect domains of consistent epigenetic patterns from ChIP-seq data, providing new insights into the role of epigenetics in long- range gene regulation. In Chapter 2, we expand our model to investigate the genome-wide patterns of histone modifications in multiple human cell lines. -

A Bacterial Protein Targets the BAHD1 Chromatin Complex to Stimulate Type III Interferon Response
A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response Alice Lebreton, Goran Lakisic, Viviana Job, Lauriane Fritsch, To Nam Tham, Ana Camejo, Pierre-Jean Matteï, Béatrice Regnault, Marie-Anne Nahori, Didier Cabanes, et al. To cite this version: Alice Lebreton, Goran Lakisic, Viviana Job, Lauriane Fritsch, To Nam Tham, et al.. A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science, American Association for the Advancement of Science, 2011, 331 (6022), pp.1319-21. 10.1126/sci- ence.1200120. cea-00819299 HAL Id: cea-00819299 https://hal-cea.archives-ouvertes.fr/cea-00819299 Submitted on 26 Jul 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Lebreton et al. Science 2011 doi:10.1126/science.1200120 A Bacterial Protein Targets the BAHD1 Chromatin Complex to Stimulate Type III Interferon Response Alice Lebreton1,2,3, Goran Lakisic4, Viviana Job5, Lauriane Fritsch6, To Nam Tham1,2,3, Ana Camejo7, Pierre-Jean Matteï5, Béatrice Regnault8, Marie-Anne Nahori1,2,3, Didier Cabanes7, Alexis Gautreau4, Slimane Ait-Si-Ali6, Andréa Dessen5, Pascale Cossart1,2,3* and Hélène Bierne1,2,3* 1. -

Download Special Issue
BioMed Research International Novel Bioinformatics Approaches for Analysis of High-Throughput Biological Data Guest Editors: Julia Tzu-Ya Weng, Li-Ching Wu, Wen-Chi Chang, Tzu-Hao Chang, Tatsuya Akutsu, and Tzong-Yi Lee Novel Bioinformatics Approaches for Analysis of High-Throughput Biological Data BioMed Research International Novel Bioinformatics Approaches for Analysis of High-Throughput Biological Data Guest Editors: Julia Tzu-Ya Weng, Li-Ching Wu, Wen-Chi Chang, Tzu-Hao Chang, Tatsuya Akutsu, and Tzong-Yi Lee Copyright © 2014 Hindawi Publishing Corporation. All rights reserved. This is a special issue published in “BioMed Research International.” All articles are open access articles distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Contents Novel Bioinformatics Approaches for Analysis of High-Throughput Biological Data,JuliaTzu-YaWeng, Li-Ching Wu, Wen-Chi Chang, Tzu-Hao Chang, Tatsuya Akutsu, and Tzong-Yi Lee Volume2014,ArticleID814092,3pages Evolution of Network Biomarkers from Early to Late Stage Bladder Cancer Samples,Yung-HaoWong, Cheng-Wei Li, and Bor-Sen Chen Volume 2014, Article ID 159078, 23 pages MicroRNA Expression Profiling Altered by Variant Dosage of Radiation Exposure,Kuei-FangLee, Yi-Cheng Chen, Paul Wei-Che Hsu, Ingrid Y. Liu, and Lawrence Shih-Hsin Wu Volume2014,ArticleID456323,10pages EXIA2: Web Server of Accurate and Rapid Protein Catalytic Residue Prediction, Chih-Hao Lu, Chin-Sheng -

Supplemental Data.Pdf
Supplementary material -Table of content Supplementary Figures (Fig 1- Fig 6) Supplementary Tables (1-13) Lists of genes belonging to distinct biological processes identified by GREAT analyses to be significantly enriched with UBTF1/2-bound genes Supplementary Table 14 List of the common UBTF1/2 bound genes within +/- 2kb of their TSSs in NIH3T3 and HMECs. Supplementary Table 15 List of gene identified by microarray expression analysis to be differentially regulated following UBTF1/2 knockdown by siRNA Supplementary Table 16 List of UBTF1/2 binding regions overlapping with histone genes in NIH3T3 cells Supplementary Table 17 List of UBTF1/2 binding regions overlapping with histone genes in HMEC Supplementary Table 18 Sequences of short interfering RNA oligonucleotides Supplementary Table 19 qPCR primer sequences for qChIP experiments Supplementary Table 20 qPCR primer sequences for reverse transcription-qPCR Supplementary Table 21 Sequences of primers used in CHART-PCR Supplementary Methods Supplementary Fig 1. (A) ChIP-seq analysis of UBTF1/2 and Pol I (POLR1A) binding across mouse rDNA. UBTF1/2 is enriched at the enhancer and promoter regions and along the entire transcribed portions of rDNA with little if any enrichment in the intergenic spacer (IGS), which separates the rDNA repeats. This enrichment coincides with the distribution of the largest subunit of Pol I (POLR1A) across the rDNA. All sequencing reads were mapped to the published complete sequence of the mouse rDNA repeat (Gene bank accession number: BK000964). The graph represents the frequency of ribosomal sequences enriched in UBTF1/2 and Pol I-ChIPed DNA expressed as fold change over those of input genomic DNA. -

Tsrna Signatures in Cancer
tsRNA signatures in cancer Veronica Balattia, Giovanni Nigitaa,1, Dario Venezianoa,1, Alessandra Druscoa, Gary S. Steinb,c, Terri L. Messierb,c, Nicholas H. Farinab,c, Jane B. Lianb,c, Luisa Tomaselloa, Chang-gong Liud, Alexey Palamarchuka, Jonathan R. Harte, Catherine Belle, Mariantonia Carosif, Edoardo Pescarmonaf, Letizia Perracchiof, Maria Diodorof, Andrea Russof, Anna Antenuccif, Paolo Viscaf, Antonio Ciardig, Curtis C. Harrish, Peter K. Vogte, Yuri Pekarskya,2, and Carlo M. Crocea,2 aDepartment of Cancer Biology and Medical Genetics, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; bDepartment of Biochemistry, University of Vermont College of Medicine, Burlington, VT 05405; cUniversity of Vermont Cancer Center, College of Medicine, Burlington, VT 05405; dMD Anderson Cancer Center, Houston, TX 77030; eDepartment of Molecular Medicine, The Scripps Research Institute, La Jolla, CA 92037; fIstituto di Ricovero e Cura a Carattere Scientifico, Regina Elena National Cancer Institute, 00144 Rome, Italy; gUniversita’ Di Roma La Sapienza, 00185 Rome, Italy; and hLaboratory of Human Carcinogenesis, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892 Contributed by Carlo M. Croce, June 13, 2017 (sent for review April 26, 2017; reviewed by Riccardo Dalla-Favera and Philip N. Tsichlis) Small, noncoding RNAs are short untranslated RNA molecules, some these molecules, which we defined as single-stranded small of which have been associated with cancer development. Recently RNAs, 16–48 nt long, ending with a stretch of four Ts (4). When we showed that a class of small RNAs generated during the matu- tsRNAs accumulate in the nucleus, they can be exported, sug- ration process of tRNAs (tRNA-derived small RNAs, hereafter gesting that tsRNAs could regulate gene expression at different “tsRNAs”) is dysregulated in cancer. -
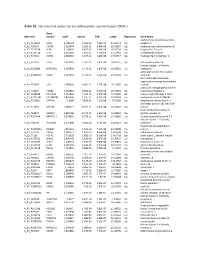
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

The Pdx1 Bound Swi/Snf Chromatin Remodeling Complex Regulates Pancreatic Progenitor Cell Proliferation and Mature Islet Β Cell
Page 1 of 125 Diabetes The Pdx1 bound Swi/Snf chromatin remodeling complex regulates pancreatic progenitor cell proliferation and mature islet β cell function Jason M. Spaeth1,2, Jin-Hua Liu1, Daniel Peters3, Min Guo1, Anna B. Osipovich1, Fardin Mohammadi3, Nilotpal Roy4, Anil Bhushan4, Mark A. Magnuson1, Matthias Hebrok4, Christopher V. E. Wright3, Roland Stein1,5 1 Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 2 Present address: Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN 3 Department of Cell and Developmental Biology, Vanderbilt University, Nashville, TN 4 Diabetes Center, Department of Medicine, UCSF, San Francisco, California 5 Corresponding author: [email protected]; (615)322-7026 1 Diabetes Publish Ahead of Print, published online June 14, 2019 Diabetes Page 2 of 125 Abstract Transcription factors positively and/or negatively impact gene expression by recruiting coregulatory factors, which interact through protein-protein binding. Here we demonstrate that mouse pancreas size and islet β cell function are controlled by the ATP-dependent Swi/Snf chromatin remodeling coregulatory complex that physically associates with Pdx1, a diabetes- linked transcription factor essential to pancreatic morphogenesis and adult islet-cell function and maintenance. Early embryonic deletion of just the Swi/Snf Brg1 ATPase subunit reduced multipotent pancreatic progenitor cell proliferation and resulted in pancreas hypoplasia. In contrast, removal of both Swi/Snf ATPase subunits, Brg1 and Brm, was necessary to compromise adult islet β cell activity, which included whole animal glucose intolerance, hyperglycemia and impaired insulin secretion. Notably, lineage-tracing analysis revealed Swi/Snf-deficient β cells lost the ability to produce the mRNAs for insulin and other key metabolic genes without effecting the expression of many essential islet-enriched transcription factors. -

NIH Public Access Author Manuscript Nat Genet
NIH Public Access Author Manuscript Nat Genet. Author manuscript; available in PMC 2011 December 14. NIH-PA Author Manuscript Published in final edited form as: Nat Genet. 2010 January ; 42(1): 53±61. doi:10.1038/ng.496. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells Stefan Schoenfelder1,6, Tom Sexton1,5,6, Lyubomira Chakalova1,5,6, Nathan F Cope1, Alice Horton1, Simon Andrews2, Sreenivasulu Kurukuti1,5, Jennifer A Mitchell1,5, David Umlauf1,5, Daniela S Dimitrova1, Christopher H Eskiw1, Yanquan Luo3, Chia-Lin Wei3, Yijun Ruan3, James J Bieker4, and Peter Fraser1 1Laboratory of Chromatin and Gene Expression, The Babraham Institute, Babraham Research Campus, Cambridge, UK 2Bioinformatics Group, The Babraham Institute, Babraham Research Campus, Cambridge, UK 3Genome Institute of Singapore, Singapore, Singapore NIH-PA Author Manuscript 4Department of Developmental and Regenerative Biology, Mount Sinai School of Medicine, New York, New York, USA Abstract The discovery of interchromosomal interactions in higher eukaryotes points to a functional interplay between genome architecture and gene expression, challenging the view of transcription as a one-dimensional process. However, the extent of interchromosomal interactions and the underlying mechanisms are unknown. Here we present the first genome-wide analysis of transcriptional interactions using the mouse globin genes in erythroid tissues. Our results show that the active globin genes associate with hundreds of other transcribed genes, revealing extensive and preferential intra- and interchromosomal transcription interactomes. We show that the transcription factor Klf1 mediates preferential co-associations of Klf1-regulated genes at a limited number of specialized transcription factories. Our results establish a new gene expression paradigm, implying that active co-regulated genes and their regulatory factors cooperate to create specialized nuclear hot spots optimized for efficient and coordinated transcriptional control. -

Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Fall 2010 Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Renuka Nayak University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Computational Biology Commons, and the Genomics Commons Recommended Citation Nayak, Renuka, "Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress" (2010). Publicly Accessible Penn Dissertations. 1559. https://repository.upenn.edu/edissertations/1559 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1559 For more information, please contact [email protected]. Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Abstract Genes interact in networks to orchestrate cellular processes. Here, we used coexpression networks based on natural variation in gene expression to study the functions and interactions of human genes. We asked how these networks change in response to stress. First, we studied human coexpression networks at baseline. We constructed networks by identifying correlations in expression levels of 8.9 million gene pairs in immortalized B cells from 295 individuals comprising three independent samples. The resulting networks allowed us to infer interactions between biological processes. We used the network to predict the functions of poorly-characterized human genes, and provided some experimental support. Examining genes implicated in disease, we found that IFIH1, a diabetes susceptibility gene, interacts with YES1, which affects glucose transport. Genes predisposing to the same diseases are clustered non-randomly in the network, suggesting that the network may be used to identify candidate genes that influence disease susceptibility.