Genetic and Clinical Features of SCN8A Developmental and Epileptic Encephalopathy T
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

S Values in Accordance with Soczewiński-Wachtmeisters Equation
S values in accordance with Soczewiński-Wachtmeisters equation S value from Antiparasitic drugs: Soczewiński’s Metronidazole Ornidazole Secnidazole Tinidazole Equation* S(m) 1.614 2.161 1.921 1.911 S(a) 1.634 2.159 1.887 1.923 S value from Antihypertensive drugs: Soczewiński’s Nilvadipine Felodipine Isradipine Lacidipine equation 4.129 4.597 3.741 5.349 S(m) S(a) 5.330 4.841 4.715 5.690 S value from Non-steroidal anti-inflammatory drugs (NSAIDs): Soczewiński’s Mefenamic Indomethacin Nabumetone Phenylbutazone Carprofen Ketoprofen Flurbiprofen equation acid 2.879 2.773 3.456 2.682 2.854 2.139 2.568 S(m) 3.317 3.442 3.910 2.404 3.394 1.968 2.010 S(a) *where: S(m) – S is the slope of the regression curie in accordance with Soczewiński-Wachtmeisters equation using methanol-water mobile phase S(a) – S is the slope of the regression curie in accordance with Soczewiński-Wachtmeisters equation using acetone-water mobile phase First group of drugs (antiparasitic drugs) 1. Metronidazole (2-Methyl-5-nitroimidazole-1-ethanol) 2. Ornidazole (1-(3-Chloro-2-hydroxypropyl)-2-methyl-5-nitroimidazole) 3. Secnidazole (1-(2-methyl-5-nitro-1H-imidazol-1-yl) propan-2ol, 1-(2Hydroxypropyl)-2-methyl-5- nitroimidazole) 4. Tinidazole (1-[2-(Ethylsulfonyl)ethyl]-2-methyl-5-nitroimidazole) Second group of drugs (antihypertensive drugs) 1. Nilvadipine (2-Cyano-1,4-dihydro-6-methyl-4-(3-nitrophenyl)-3,5-pyridinedicarboxylic acid 3- methyl 5-(1-methylethyl) ester, 5-Isopropyl-3-methyl-2-cyano-1,4-dihydro-6-methyl-4-(m- nitrophenyl)-3,5-pyridinedicarboxylate, FK-235, FR-34235, Isopropyl 6-cyano-5-methoxycarbonyl-2- methyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3-carboxylate) 2. -

Dorset Medicines Advisory Group
DORSET CARDIOLOGY WORKING GROUP GUIDELINE FOR CALCIUM CHANNEL BLOCKERS IN HYPERTENSION SUMMARY The pan-Dorset cardiology working group continues to recommend the use of amlodipine (a third generation dihydropyridine calcium-channel blocker) as first choice calcium channel blocker on the pan-Dorset formulary for hypertension. Lercanidipine is second choice, lacidipine third choice and felodipine is fourth choice. This is due to preferable side effect profiles in terms of ankle oedema and relative costs of the preparations. Note: where angina is the primary indication or is a co-morbidity prescribers must check against the specific product characteristics (SPC) for an individual drug to confirm this is a licensed indication. N.B. Lacidipine and lercandipine are only licensed for use in hypertension. Chapter 02.06.02 CCBs section of the Formulary has undergone an evidence-based review. A comprehensive literature search was carried out on NHS Evidence, Medline, EMBASE, Cochrane Database, and UK Duets. This was for recent reviews or meta-analyses on calcium channel blockers from 2009 onwards (comparative efficacy and side effects) and randomised controlled trials (RCTs). REVIEW BACKGROUND Very little good quality evidence exists. No reviews, meta-analyses or RCTs were found covering all calcium channel blockers currently on the formulary. Another limitation was difficulty obtaining full text original papers for some of the references therefore having to use those from more obscure journals instead. Some discrepancies exist between classification of generations of dihydropyridine CCBs, depending upon the year of publication of the reference/authors’ interpretation. Dihydropyridine (DHP) CCBs tend to be more potent vasodilators than non-dihydropyridine (non-DHP) CCBs (diltiazem, verapamil), but the latter have greater inotropic effects. -

Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options
1521-0081/72/3/606–638$35.00 https://doi.org/10.1124/pr.120.019539 PHARMACOLOGICAL REVIEWS Pharmacol Rev 72:606–638, July 2020 Copyright © 2020 by The Author(s) This is an open access article distributed under the CC BY-NC Attribution 4.0 International license. ASSOCIATE EDITOR: ERIC L. BARKER Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options Wolfgang Löscher, Heidrun Potschka, Sanjay M. Sisodiya, and Annamaria Vezzani Department of Pharmacology, Toxicology, and Pharmacy, University of Veterinary Medicine, Hannover, Germany (W.L.); Center for Systems Neuroscience, Hannover, Germany (W.L.); Institute of Pharmacology, Toxicology and Pharmacy, Ludwig-Maximilians-University, Munich, Germany (H.P.); Department of Clinical and Experimental Epilepsy, UCL Queen Square Institute of Neurology, London, United Kingdom (S.S); and Department of Neuroscience, Mario Negri Institute for Pharmacological Research Istituto di Ricovero e Cura a Carattere Scientifico, Milano, Italy (A.V.) Abstract. ....................................................................................607 Significance Statement ......................................................................607 I. Introduction. ..............................................................................607 II. Pharmacology of Antiseizure Drugs ..........................................................608 III. In Vivo and In Vitro Models of Drug Resistance..............................................610 A. General Aspects . ........................................................................610 -

Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Cr
Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Crizotinib (PF-02341066) 1 4 55 Docetaxel 1 5 98 Anastrozole 1 6 25 Cladribine 1 7 23 Methotrexate 1 8 -187 Letrozole 1 9 65 Entecavir Hydrate 1 10 48 Roxadustat (FG-4592) 1 11 19 Imatinib Mesylate (STI571) 1 12 0 Sunitinib Malate 1 13 34 Vismodegib (GDC-0449) 1 14 64 Paclitaxel 1 15 89 Aprepitant 1 16 94 Decitabine 1 17 -79 Bendamustine HCl 1 18 19 Temozolomide 1 19 -111 Nepafenac 1 20 24 Nintedanib (BIBF 1120) 1 21 -43 Lapatinib (GW-572016) Ditosylate 1 22 88 Temsirolimus (CCI-779, NSC 683864) 1 23 96 Belinostat (PXD101) 1 24 46 Capecitabine 1 25 19 Bicalutamide 1 26 83 Dutasteride 1 27 68 Epirubicin HCl 1 28 -59 Tamoxifen 1 29 30 Rufinamide 1 30 96 Afatinib (BIBW2992) 1 31 -54 Lenalidomide (CC-5013) 1 32 19 Vorinostat (SAHA, MK0683) 1 33 38 Rucaparib (AG-014699,PF-01367338) phosphate1 34 14 Lenvatinib (E7080) 1 35 80 Fulvestrant 1 36 76 Melatonin 1 37 15 Etoposide 1 38 -69 Vincristine sulfate 1 39 61 Posaconazole 1 40 97 Bortezomib (PS-341) 1 41 71 Panobinostat (LBH589) 1 42 41 Entinostat (MS-275) 1 43 26 Cabozantinib (XL184, BMS-907351) 1 44 79 Valproic acid sodium salt (Sodium valproate) 1 45 7 Raltitrexed 1 46 39 Bisoprolol fumarate 1 47 -23 Raloxifene HCl 1 48 97 Agomelatine 1 49 35 Prasugrel 1 50 -24 Bosutinib (SKI-606) 1 51 85 Nilotinib (AMN-107) 1 52 99 Enzastaurin (LY317615) 1 53 -12 Everolimus (RAD001) 1 54 94 Regorafenib (BAY 73-4506) 1 55 24 Thalidomide 1 56 40 Tivozanib (AV-951) 1 57 86 Fludarabine -

Antioxidant Effects of Calcium Antagonists Rat Myocardial
223 Antioxidant Effects of Calcium Antagonists ['Ii] Rat Myocardial Membrane Lipid Peroxidation Hitoshi Sugawara, Katsuyuki Tobise, and Kenjiro Kikuchi We studied the antioxidant effects of nine calcium antagonists (nisoldipine, benidipine, nilvadipine, felo- dipine, nicardipine; nitrendipine, nifedipine, verapamil, and diltiazem) by means of rat myocardial membrane lipid peroxidation with a nonenzymatic active oxygen-generating system (DHF/FeC13-ADP). The order of antioxidant potency of these agents was nilvadipine > nisoldipine > felodipine > nicardipine > verapamil > benidipine. Their IC50 values (,uM) were 25.1, 28.2, 42.0, 150.0, 266.1, and 420.0, re- spectively. In contrast, nitrendipine, nifedipine, and diltiazem had little inhibitory effect on lipid peroxi- dation.These six calcium antagonists could be divided into four types on the basis of their antioxidant mechanisms. Nilvadipine, nisoldipine, and verapamil, which showed antioxidant effects both before and after the addition of active oxygen, and reduced the dihydroxyfumarate (DHF) auto-oxidation rate, were chain-breaking and preventive antioxidants. Felodipine, which showed antioxidant effects both before and after exposure to active oxygen and increased the DHF auto-oxidation rate, was only a chain-break- ing antioxidant. Nicardipine, which showed an antioxidant effect only before exposure to active oxygen and reduced the DHF auto-oxidation rate, was mainly a preventive antioxidant. Benidipine, which showed an antioxidant effect only before exposure to active oxygen and had no appreciable effect on the DHF auto-oxidation rate, could interrupt the chain reaction of lipid peroxidation at the initial step alone. Although these results suggest that the antioxidant properties of some calcium antagonists may be beneficial clinically in protecting against cellular damage caused by lipid peroxidation, further studies are required to establish the antioxidant effects of these agents in vivo. -

Download Leaflet View the Patient Leaflet in PDF Format
Package leaflet: Information for the patient Carbamazepine SUN 100 mg/5 ml Oral Suspension carbamazepine Read all of this leaflet carefully before you take this medicine because it contains important information for you. − Keep this leaflet. You may need to read it again. − If you have further questions, ask your doctor or pharmacist. − This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. − If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet 1. What Carbamazepine SUN is and what it is used for 2. What you need to know before you take Carbamazepine SUN 3. How to take Carbamazepine SUN 4. Possible side effects 5. How to store Carbamazepine SUN 6. Contents of the pack and other information 1. What Carbamazepine SUN is and what it is used for Carbamazepine SUN is a pale orange suspension that contains carbamazepine, the active ingredient. Carbamazepine SUN is an anti-convulsant medicine (prevents fits), it can also modify some types of pain and can control mood disorders. Carbamazepine SUN is used - to treat some forms of epilepsy - to treat a painful condition of the face called trigeminal neuralgia - to help control serious mood disorders when some other medicines don't work. 2. What you need to know before you take Carbamazepine SUN Do not take Carbamazepine SUN - if you are allergic to carbamazepine or similar drugs such as oxcarbazepine (Trileptal), or to any of a related group of drugs known as tricyclic antidepressants (such as amitriptyline or imipramine) or any of the other ingredients of this medicine (listed in section 6). -

The Following Full Text Is a Publisher's Version
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/137509 Please be advised that this information was generated on 2021-09-25 and may be subject to change. BMJ Open 2014;4:e006364 doi:10.1136/bmjopen-2014-006364 Geriatric medicine Protocol NILVAD protocol: a European multicentre double-blind placebo-controlled trial of nilvadipine in mild- to-moderate Alzheimer's disease Brian Lawlor1, Sean Kennelly1, Sarah O'Dwyer1, Fiona Cregg2, Cathal Walsh2, Robert Coen1, Rose Anne Kenny1, Robert Howard3, Caroline Murphy3, Jessica Adams3, Leslie Daly4, Ricardo Segurado4, Siobhan Gaynor5, Fiona Crawford6, Michael Mullan6, Ugo Lucca7, Rita Banzi7, Florence Pasquier8, Laetitia Breuilh8, Matthias Riepe9, Janos Kalman10, Anders Wallin11, Anne Borjesson11, William Molloy12, Magda Tsolaki13, Marcel Olde Rikkert14 + Author Affiliations 1Mercer's Institute for Research on Ageing, St. James's Hospital, Dublin, Ireland 2Trinity College Dublin (TCD), Dublin, Ireland 3King's College London (KCL), London, UK 4University College Dublin (UCD), Dublin, Ireland 5Molecular Medicine Ireland (MMI), Dublin, Ireland 6Archer Pharmaceuticals Inc, 2040 Whitefield Avenue, Sarasota, Florida, USA 7IRCCS—Istituto di Ricerche Farmacologiche “Mario Negri” (IRFMN), Milan, Italy 8Centre Hospitalier Regional et Universitaire de Lille (CHRU- LILLE), Lille, France 9Universitaet Ulm, (UULM), Ulm, Germany 10Szegedi Tudomanyegyetem -
PDF-Document
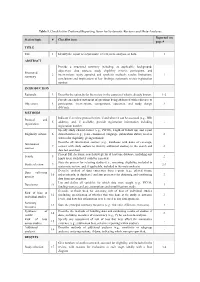
Table 1. Checklist for Preferred Reporting Items for Systematic Reviews and Meta-Analyses. Reported on Section/topic # Checklist item page # TITLE Title 1 Identify the report as a systematic review, meta-analysis, or both. 1 ABSTRACT Provide a structured summary including, as applicable: background; objectives; data sources; study eligibility criteria, participants, and Structured 2 interventions; study appraisal and synthesis methods; results; limitations; 1 summary conclusions and implications of key findings; systematic review registration number. INTRODUCTION Rationale 3 Describe the rationale for the review in the context of what is already known. 1-2 Provide an explicit statement of questions being addressed with reference to Objectives 4 participants, interventions, comparisons, outcomes, and study design 2 (PICOS). METHODS Indicate if a review protocol exists, if and where it can be accessed (e.g., Web Protocol and 5 address), and, if available, provide registration information including - registration registration number. Specify study characteristics (e.g., PICOS, length of follow-up) and report Eligibility criteria 6 characteristics (e.g., years considered, language, publication status) used as 2 criteria for eligibility, giving rationale. Describe all information sources (e.g., databases with dates of coverage, Information 7 contact with study authors to identify additional studies) in the search and 2 sources date last searched. Present full electronic search strategy for at least one database, including any Search 8 2 limits used, such that it could be repeated. State the process for selecting studies (i.e., screening, eligibility, included in Study selection 9 2-3 systematic review, and, if applicable, included in the meta-analysis). Describe method of data extraction from reports (e.g., piloted forms, Data collection 10 independently, in duplicate) and any processes for obtaining and confirming 3 process data from investigators. -

Summary of Product Characteristics
Health Products Regulatory Authority Summary of Product Characteristics 1 NAME OF THE MEDICINAL PRODUCT Nivadil 16mg Prolonged release capsules 2 QUALITATIVE AND QUANTITATIVE COMPOSITION Each capsule contains Nilvadipine 16.0 mg For the full list of excipents, see section 6.1. 3 PHARMACEUTICAL FORM Prolonged release capsule, hard. (Prolonged release capsule) Hard gelatin capsules with an opaque brown cap (overprinted with "NV16" in white) and an opaque brown/red body containing yellow round pellets. 4 CLINICAL PARTICULARS 4.1 Therapeutic Indications Nivadil is indicated for the treatment of essential hypertension. 4.2 Posology and method of administration For oral administration. The prolonged release capsule should be swallowed whole, with a little liquid, in the morning. It may be taken after breakfast. Adults The recommended dose is 1 Nivadil 8 mg prolonged release capsule per day in the morning as a starting dose. If after 2 - 4 weeks of therapy an adequate anti -hypertensive effect is not achieved, a daily dose of 16 mg nilvadipine (2 x 8 mg Nivadil prolonged release capsules, or 1 x 16 mg Nivadil prolonged release capsule, in the morning) may improve the blood pressure response. Renal impairment: No dosage adjustment is required in mild to moderate renal insufficiency. Nivadil should not however be used in patients with severe renal insufficiency. Hepatic impairment: In patients with cirrhosis of the liver, due to diminished first -pass effect, the bioavailability is increased by a factor of 2 to 3. The currently available data lead to the recommendation that a daily dose of 1 x 8 mg nilvadipine (equivalent to 1 Nivadil 8 mg prolonged release capsule) may only be exceeded under close monitoring in such patients. -

Prescription Medications, Drugs, Herbs & Chemicals Associated With
Prescription Medications, Drugs, Herbs & Chemicals Associated with Tinnitus American Tinnitus Association Prescription Medications, Drugs, Herbs & Chemicals Associated with Tinnitus All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form, or by any means, without the prior written permission of the American Tinnitus Association. ©2013 American Tinnitus Association Prescription Medications, Drugs, Herbs & Chemicals Associated with Tinnitus American Tinnitus Association This document is to be utilized as a conversation tool with your health care provider and is by no means a “complete” listing. Anyone reading this list of ototoxic drugs is strongly advised NOT to discontinue taking any prescribed medication without first contacting the prescribing physician. Just because a drug is listed does not mean that you will automatically get tinnitus, or exacerbate exisiting tinnitus, if you take it. A few will, but many will not. Whether or not you eperience tinnitus after taking one of the listed drugs or herbals, or after being exposed to one of the listed chemicals, depends on many factors ‐ such as your own body chemistry, your sensitivity to drugs, the dose you take, or the length of time you take the drug. It is important to note that there may be drugs NOT listed here that could still cause tinnitus. Although this list is one of the most complete listings of drugs associated with tinnitus, no list of this kind can ever be totally complete – therefore use it as a guide and resource, but do not take it as the final word. The drug brand name is italicized and is followed by the generic drug name in bold. -

Reduces Ischemic Brain Injury in Rats
Nilvadipine, a New Calcium Channel Blocker, Reduces Ischemic Brain Injury in Rats Shoji Takakura, Teruo Susumu, Hisashi Satoh, Jo Mori, Akihiko Shiino' and Jyoji Handa' Department of Pharmacology, Product Development Laboratories, Fujisawa Pharmaceutical Co., Ltd., 2-1-6, Kashima, Osaka 532, Japan 'Department of Neurosurgery, Shiga University of Medical Sciences, Seta, Ohtsu 520-21, Japan Received February 7, 1991 Accepted June 10, 1991 ABSTRACT-The effects of nilvadipine, a dihydropyridine type calcium channel blocker, on cerebral infarction induced by focal brain ischemia was studied in rats. The area of infarction was measured 24 hr after occlusion of the middle cerebral artery (MCA) in spontaneously hypertensive rats using triphenyltetrazolium chloride. Nilvadipine, given immediately after MCA occlusion, reduced the area of infarction significantly at doses of 0.32 mg/kg (i.p.) and 3.2, 10 and 32 mg/kg (p.o.). Nicardi pine suppressed the area of infarction at a dose of 32 mg/kg (p.o.). The results sug gest that nilvadipine is effective against ischemic brain injury. Ischemic cerebrovascular disease remains ample, it had no activity on the general be the most prevalent neurologic disorder. Cal havior in conscious dogs (T. Ono et al., un cium channel blockers have received attention published data). These results prompted us to as possible neuroprotective agents in ischemia study the effect of nilvadipine on experimental both because of their potent cerebral vasodila cerebral infarction in rats. In this study, we tory activity (1, 2) and because they may pro examined the effects of nilvadipine on the size tect neurons by preventing the accumulation of the infarction in the MCA occluded rat. -

Drugs and Scaffold That Inhibit Cytochrome P450 27A1 (CYP27A1) in Vitro and in Vivo
Molecular Pharmacology Fast Forward. Published on November 30, 2017 as DOI: 10.1124/mol.117.110742 This article has not been copyedited and formatted. The final version may differ from this version. MOL #110742 Downloaded from Drugs and Scaffold that Inhibit Cytochrome P450 27A1 (CYP27A1) in Vitro and in Vivo molpharm.aspetjournals.org Morrie Lam, Natalia Mast, and Irina A. Pikuleva Department of Ophthalmology and Visual Sciences, Case Western Reserve University, at ASPET Journals on September 25, 2021 Cleveland, Ohio 1 Molecular Pharmacology Fast Forward. Published on November 30, 2017 as DOI: 10.1124/mol.117.110742 This article has not been copyedited and formatted. The final version may differ from this version. MOL #110742 a) Running title: CYP27A1 Inhibition by Drugs b) Corresponding author: Irina A. Pikuleva, Department of Ophthalmology and Visual Sciences, Case Western Reserve University, 2085 Adelbert Rd., Cleveland, OH 44106. E-mail: [email protected]. Downloaded from c) 30 text pages 1 table 4 figures molpharm.aspetjournals.org 40 references 249 words in the Abstract 642 words in the Introduction at ASPET Journals on September 25, 2021 1,174 words in the Discussion d) Non-standard abbreviations: 27HC, 27-hydroxycholesterol; CTX, cerebrotendinous xanthomatosis; DHP, 1,4-dihydro-pyridine; ER, estrogen receptor; FDA, Food and Drug Administration, KPi, potassium phosphate. 2 Molecular Pharmacology Fast Forward. Published on November 30, 2017 as DOI: 10.1124/mol.117.110742 This article has not been copyedited and formatted. The final version may differ from this version. MOL #110742 ABSTRACT Cytochrome P450 27A1 (CYP27A1) is a ubiquitous enzyme that hydroxylates cholesterol and other sterols.