An Image-Based Data-Driven Analysis of Cellular Architecture in a Developing Tissue Jonas Hartmann1*, Mie Wong2, Elisa Gallo1,2,3, Darren Gilmour2*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Management of Large Sets of Image Data Capture, Databases, Image Processing, Storage, Visualization Karol Kozak
Management of large sets of image data Capture, Databases, Image Processing, Storage, Visualization Karol Kozak Download free books at Karol Kozak Management of large sets of image data Capture, Databases, Image Processing, Storage, Visualization Download free eBooks at bookboon.com 2 Management of large sets of image data: Capture, Databases, Image Processing, Storage, Visualization 1st edition © 2014 Karol Kozak & bookboon.com ISBN 978-87-403-0726-9 Download free eBooks at bookboon.com 3 Management of large sets of image data Contents Contents 1 Digital image 6 2 History of digital imaging 10 3 Amount of produced images – is it danger? 18 4 Digital image and privacy 20 5 Digital cameras 27 5.1 Methods of image capture 31 6 Image formats 33 7 Image Metadata – data about data 39 8 Interactive visualization (IV) 44 9 Basic of image processing 49 Download free eBooks at bookboon.com 4 Click on the ad to read more Management of large sets of image data Contents 10 Image Processing software 62 11 Image management and image databases 79 12 Operating system (os) and images 97 13 Graphics processing unit (GPU) 100 14 Storage and archive 101 15 Images in different disciplines 109 15.1 Microscopy 109 360° 15.2 Medical imaging 114 15.3 Astronomical images 117 15.4 Industrial imaging 360° 118 thinking. 16 Selection of best digital images 120 References: thinking. 124 360° thinking . 360° thinking. Discover the truth at www.deloitte.ca/careers Discover the truth at www.deloitte.ca/careers © Deloitte & Touche LLP and affiliated entities. Discover the truth at www.deloitte.ca/careers © Deloitte & Touche LLP and affiliated entities. -

System Trends and Their Impact on Future Microprocessor Design
IBM Research System Trends and their Impact on Future Microprocessor Design Tilak Agerwala Vice President, Systems IBM Research System Trends and their Impact | MICRO 35 | Tilak Agerwala © 2002 IBM Corporation IBM Research Agenda System and application trends Impact on architecture and microarchitecture The Memory Wall Cellular architectures and IBM's Blue Gene Summary System Trends and their Impact | MICRO 35 | Tilak Agerwala © 2002 IBM Corporation IBM Research TRENDSTRENDS Microprocessors < 10 GHz in systems 64-256 Way SMP 65-45nm, Copper, Highest performance SOI Best MP Scalability 1-2 GHz Leading edge process technology 4-8 Way SMP RAS, virtualization ~100nm technology 10+ of GHz 4-8 Way SMP SMP/Large 65-45nm, Copper, SOI Systems Highest Frequency Cost and power Low GHz sensitive Leading edge process Uniprocessor technology Desktop ~100nm technology 2-4 GHz, Uniproc, Component-based and Game ~100nm, Copper, SOI Consoles Lowest Power / Lowest Multi MHz cost designs SoC capable Embedded Uniprocessor ASIC / Foundry ~100-200nm technologies Systems technology System Trends and their Impact | MICRO 35 | Tilak Agerwala © 2002 IBM Corporation IBM Research TRENDSTRENDS Large system application trends Traditional commercial applications Databases, transaction processing, business apps like payroll etc. The internet has driven the growth of new commercial applications New life sciences applications are commercial and high-growth Drug discovery and genetic engineering research needs huge amounts of compute power (e.g. protein folding simulations) -

An Overview of the Blue Gene/L System Software Organization
An Overview of the Blue Gene/L System Software Organization George Almasi´ , Ralph Bellofatto , Jose´ Brunheroto , Calin˘ Cas¸caval , Jose´ G. ¡ Castanos˜ , Luis Ceze , Paul Crumley , C. Christopher Erway , Joseph Gagliano , Derek Lieber , Xavier Martorell , Jose´ E. Moreira , Alda Sanomiya , and Karin ¡ Strauss ¢ IBM Thomas J. Watson Research Center Yorktown Heights, NY 10598-0218 £ gheorghe,ralphbel,brunhe,cascaval,castanos,pgc,erway, jgaglia,lieber,xavim,jmoreira,sanomiya ¤ @us.ibm.com ¥ Department of Computer Science University of Illinois at Urbana-Champaign Urabana, IL 61801 £ luisceze,kstrauss ¤ @uiuc.edu Abstract. The Blue Gene/L supercomputer will use system-on-a-chip integra- tion and a highly scalable cellular architecture. With 65,536 compute nodes, Blue Gene/L represents a new level of complexity for parallel system software, with specific challenges in the areas of scalability, maintenance and usability. In this paper we present our vision of a software architecture that faces up to these challenges, and the simulation framework that we have used for our experiments. 1 Introduction In November 2001 IBM announced a partnership with Lawrence Livermore National Laboratory to build the Blue Gene/L (BG/L) supercomputer, a 65,536-node machine de- signed around embedded PowerPC processors. Through the use of system-on-a-chip in- tegration [10], coupled with a highly scalable cellular architecture, Blue Gene/L will de- liver 180 or 360 Teraflops of peak computing power, depending on the utilization mode. Blue Gene/L represents a new level of scalability for parallel systems. Whereas existing large scale systems range in size from hundreds (ASCI White [2], Earth Simulator [4]) to a few thousands (Cplant [3], ASCI Red [1]) of compute nodes, Blue Gene/L makes a jump of almost two orders of magnitude. -

Road Lane Detection for Android
MASARYK UNIVERSITY FACULTY}w¡¢£¤¥¦§¨ OF I !"#$%&'()+,-./012345<yA|NFORMATICS Road lane detection for Android BACHELOR THESIS Jakub Medvecký-Heretik Brno, spring 2014 Declaration I do hereby declare, that this paper is my original authorial work, which I have worked on by my own. All sources, references and liter- ature used or excerpted during elaboration of this work are properly cited and listed in complete reference to the due source. Jakub Medvecký-Heretik Advisor: Mgr. Dušan Klinec Acknowledgement I would like to thank my supervisor, Mgr. Dušan Klinec for his con- tinual support and advice throughout this work. I would also like to thank my family and friends for their support. Abstract The aim of this bachelor thesis was to analyze the possibilities of im- age processing techniques on the Android mobile platform for the purposes of a computer vision application focusing on the problem of road lane detection. Utilizing the results of the analysis an appli- cation was created, which detects lines and circles in an image in real time using various approaches to segmentation and different imple- mentations of the Hough transform. The application makes use of the OpenCV library and the results achieved with its help are com- pared to the results achieved with my own implementation. Keywords computer vision, road lane detection, circle detection, image process- ing, segmentation, Android, OpenCV, Hough transform Contents 1 Introduction ............................ 3 2 Computer vision ......................... 5 2.1 Image processing ...................... 5 2.2 Techniques .......................... 6 2.3 Existing libraries ...................... 6 2.3.1 OpenCV . 6 2.3.2 VXL . 7 2.3.3 ImageJ . -

Cellular Wave Computers and CNN Technology – a Soc Architecture with Xk Processors and Sensor Arrays*
Cellular Wave Computers and CNN Technology – a SoC architecture with xK processors and sensor arrays* Tamás ROSKA1 Fellow IEEE 1. Introduction and the main theses 1.1 Scenario: Architectural lessons from the trends in manufacturing billion component devices when crossing the threshold of 100 nm feature size Preliminary proposition: The nature of fabrication technology, the nature and type of data to be processed, and the nature and type of events to be detected or „computed” will determine the architecture, the elementary instructions, and the type of algorithms needed, hence also the complexity of the solution. In view of this proposition, let us list a few key features of the electronic technology of Today and its consequences.. (i) Convergence of CMOS, NANO and OPTICAL technologies towards a cellular architecture with short and sparse wires CMOS chips: * Processors: K or M transistors on an M or G transistor die =>K processors /chip * Wires: at 180 nm or below, gate delay is smaller than wire delay NANO processors and sensors: * Mainly 2D organization of cells integrating processing and sensing * Interactions mainly with the neighbours OPTICAL devices: * parallel processing * optical correlators * VCSELs and programable SLMs, Hence the architecture should be characterized by * 2 D layers (or a layered 3D) * Cellular architecture with * mainly local and /or regular sparse wireing leading via Î a Cellular Nonlinear Network (CNN) Dynamics 1 The Faculty of Information Technology and the Jedlik Laboratories of the Pázmány University, Budapest and the Computer and Automation Institute of the Hungarian Academy of Sciences, Budapest, Hungary ([email protected], www.itk.ppke.hu) * Research supported by the Office of Naval Research, Human Frontiers of Science Program, EU Future and Emergent Technologies Program, the Hungarian Academy of Sciences, and the Jedlik Laboratories of the Pázmány University, Budapest 0-7803-9254-X/05/$20.00 ©2005 IEEE. -

Performance Modelling and Optimization of Memory Access on Cellular Computer Architecture Cyclops64
Performance modelling and optimization of memory access on cellular computer architecture Cyclops64 Yanwei Niu, Ziang Hu, Kenneth Barner and Guang R. Gao Department of ECE, University of Delaware, Newark, DE, 19711, USA {niu, hu, barner, ggao}@ee.udel.edu Abstract. This paper focuses on the Cyclops64 computer architecture and presents an analytical model and performance simulation results for the preloading and loop unrolling approaches to optimize the performance of SVD (Singular Value Decomposition) benchmark. A performance model for dissecting the total execu- tion cycles is presented. The data preloading using “memcpy” or hand optimized “inline” assembly code, and the loop unrolling approach are implemented and compared with each other in terms of the total number of memory access cycles. The key idea is to preload data from offchip to onchip memory and store the data back after the computation. These approaches can reduce the total memory access cycles and can thus improve the benchmark performance significantly. 1 Introduction The design concept of computer architecture over the last two decades has been mainly on the exploitation of the instruction level parallelism, such as pipelining,VLIW or superscalar architecture. For the next generation of computer architecture, hardware threading multiprocessor is becoming more and more popular. One approach of hard- ware multithreading is called CMP (Chip MultiProcessor) approach, which proposes a single chip design that uses a collection of independent processors with less resource sharing. An example of CMP architecture design is Cyclops64 [1–5], a new architec- ture for high performance parallel computers being developed at the IBM T. J. Watson Research Center and University of Delaware. -
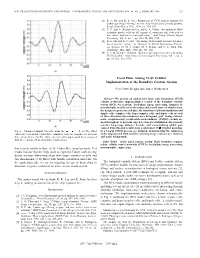
Focal-Plane Analog VLSI Cellular Implementation of the Boundary Contour System
IEEE TRANSACTIONS ON CIRCUITS AND SYSTEMS—I: FUNDAMENTAL THEORY AND APPLICATIONS, VOL. 46, NO. 2, FEBRUARY 1999 327 [6] K. F. Hui and B. E. Shi, “Robustness of CNN implementations for Gabor-type image filtering,” in Proc. Asia Pacific Conf. Circuits Systems, Seoul, South Korea, Nov. 1996, pp. 105–108. [7] C. C. Enz, F. Krummenacher, and E. A. Vittoz, “An analytical MOS transistor model valid in all regions of operation and dedicated to low-voltage and low-current applications,” Anal. Integr. Circuits Signal Processing, vol. 8, no. 1, pp. 83–114, July 1995. [8] B. E. Shi and K. F. Hui, “An analog VLSI neural network for phase based machine vision,” in Advances in Neural Information Process- ing Systems 10, M. I. Jordan, M. J. Kearns, and S. A. Solla, Eds. Cambridge, MA: MIT, 1998, pp. 726–732. [9] C. A. Mead and T. Delbruck, “Scanners for visualizing activity of analog VLSI circuitry,” Anal. Integr. Circuits Signal Processing, vol. 1, no. 2, pp. 93–106, Oct. 1991. (a) Focal-Plane Analog VLSI Cellular Implementation of the Boundary Contour System Gert Cauwenberghs and James Waskiewicz Abstract—We present an analog very large scale integration (VLSI) cellular architecture implementing a version of the boundary contour system (BCS) for real-time focal-plane image processing. Inspired by neuromorphic models across the retina and several layers of visual cortex, the design integrates in each pixel the functions of phototransduction and simple cells, complex cells, hypercomplex cells, and bipole cells in each of three directions interconnected on a hexagonal grid. Analog current- mode complementary metal–oxide–semiconductor (CMOS) circuits are used throughout to perform edge detection, local inhibition, directionally (b) selective long-range diffusive kernels, and renormalizing global gain control. -

Software-Defined Hyper-Cellular Architecture for Green and Elastic
1 Software-Defined Hyper-Cellular Architecture for Green and Elastic Wireless Access Sheng Zhou, Tao Zhao, Zhisheng Niu, and Shidong Zhou Abstract To meet the surging demand of increasing mobile Internet traffic from diverse applications while maintaining moderate energy cost, the radio access network (RAN) of cellular systems needs to take a green path into the future, and the key lies in providing elastic service to dynamic traffic demands. To achieve this, it is time to rethink RAN architectures and expect breakthroughs. In this article, we review the state-of-art literature which aims to renovate RANs from the perspectives of control-traffic decoupled air interface, cloud-based RANs, and software-defined RANs. We then propose a software- defined hyper-cellular architecture (SDHCA) that identifies a feasible way of integrating the above three trends to enable green and elastic wireless access. We further present key enabling technologies to realize SDHCA, including separation of the air interface, green base station operations, and base station functions virtualization, followed by our hardware testbed for SDHCA. Besides, we summarize several future research issues worth investigating. I. INTRODUCTION Since their birth, cellular systems have evolved from the first generation analog systems with very low data rate to today’s fourth generation (4G) systems with more than 100 Mbps capacity to end users. However, the radio access network (RAN) architecture has not experienced arXiv:1512.04935v1 [cs.NI] 15 Dec 2015 many changes: base stations (BSs) are generally deployed and operated in a distributed fashion, Sheng Zhou, Zhisheng Niu and Shidong Zhou are with Tsinghua National Laboratory for Information Science and Technology, Dept. -

An Overview on Cyclops-64 Architecture - a Status Report on the Programming Model and Software Infrastructure
An Overview on Cyclops-64 Architecture - A Status Report on the Programming Model and Software Infrastructure Guang R. Gao Endowed Distinguished Professor Electrical & Computer Engineering University of Delaware [email protected] 2007/6/14 SOS11-06-2007.ppt 1 Outline • Introduction • Multi-Core Chip Technology • IBM Cyclops-64 Architecture/Software • Cyclops-64 Programming Model and System Software • Future Directions • Summary 2007/6/14 SOS11-06-2007.ppt 2 TIPs of compute power operating on Tera-bytes of data Transistor Growth in the near future Source: Keynote talk in CGO & PPoPP 03/14/07 by Jesse Fang from Intel 2007/6/14 SOS11-06-2007.ppt 3 Outline • Introduction • Multi-Core Chip Technology • IBM Cyclops-64 Architecture/Software • Programming/Compiling for Cyclops-64 • Looking Beyond Cyclops-64 • Summary 2007/6/14 SOS11-06-2007.ppt 4 Two Types of Multi-Core Architecture Trends • Type I: Glue “heavy cores” together with minor changes • Type II: Explore the parallel architecture design space and searching for most suitable chip architecture models. 2007/6/14 SOS11-06-2007.ppt 5 Multi-Core Type II • New factors to be considered –Flops are cheap! –Memory per core is small –Cache-coherence is expensive! –On-chip bandwidth can be enormous! –Examples: Cyclops-64, and others 2007/6/14 SOS11-06-2007.ppt 6 Flops are Cheap! An example to illustrate design tradeoffs: • If fed from small, local register files: 64-bit FP – 3200 GB/s, 10 pJ/op unit – < $1/Gflop (60 mW/Gflop) (drawn to a 64-bit FPU is < 1mm^2 scale) and ~= 50pJ • If fed from global on-chip memory: Can fit over 200 on a chip. -

Virtualized Baseband Units Consolidation in Advanced Lte Networks Using Mobility- and Power-Aware Algorithms
San Jose State University SJSU ScholarWorks Master's Projects Master's Theses and Graduate Research Fall 2017 VIRTUALIZED BASEBAND UNITS CONSOLIDATION IN ADVANCED LTE NETWORKS USING MOBILITY- AND POWER-AWARE ALGORITHMS Uladzimir Karneyenka San Jose State University Follow this and additional works at: https://scholarworks.sjsu.edu/etd_projects Part of the Computer Sciences Commons Recommended Citation Karneyenka, Uladzimir, "VIRTUALIZED BASEBAND UNITS CONSOLIDATION IN ADVANCED LTE NETWORKS USING MOBILITY- AND POWER-AWARE ALGORITHMS" (2017). Master's Projects. 571. DOI: https://doi.org/10.31979/etd.sg3g-dr33 https://scholarworks.sjsu.edu/etd_projects/571 This Master's Project is brought to you for free and open access by the Master's Theses and Graduate Research at SJSU ScholarWorks. It has been accepted for inclusion in Master's Projects by an authorized administrator of SJSU ScholarWorks. For more information, please contact [email protected]. VIRTUALIZED BASEBAND UNITS CONSOLIDATION IN ADVANCED LTE NETWORKS USING MOBILITY - AND POWER -AWARE ALGORITHMS A Writing Project Report Presented to The Faculty of the Department of Computer Science San Jose State University In Partial Fulfillment Of the Requirements for the Degree Master of Science By Uladzimir Karneyenka May 2017 © 2017 Uladzimir Karneyenka ALL RIGHTS RESERVED ABSTRACT Virtualization of baseband units in Advanced Long-Term Evolution networks and a rapid performance growth of general purpose processors naturally raise the interest in resource multiplexing. The concept of resource sharing and management between virtualized instances is not new and extensively used in data centers. We adopt some of the resource management techniques to organize virtualized baseband units on a pool of hosts and investigate the behavior of the system in order to identify features which are particularly relevant to mobile environment. -

André Vilas Boas Da Costa Retscan: Efficient Fovea and Optic Disc Detection in Retinographies
Universidade do Minho Escola de Engenharia André Vilas Boas da Costa RetScan: Efficient Fovea and Optic Disc Detection in Retinographies Outubro de 2011 Universidade do Minho Escola de Engenharia Departamento de Informática André Vilas Boas da Costa RetScan: Efficient Fovea and Optic Disc Detection in Retinographies Dissertação de Mestrado Mestrado de Informática Trabalho realizado sob orientação de Alberto José Proença Outubro de 2011 Abstract The Fovea and Optic Disc are relevant anatomical eye structures to diagnose var- ious diseases. Its automatic detection can provide both a cost reduction when analysing large populations and improve the effectiveness of ophthalmologists and optometrists. This dissertation describes a methodology to automatically detect these structures and analyses a, CPU only, MATLAB implementation of this methodology. RetScan is a port to a freeware environment of this methodology, its functionality and perfor- mance are evaluated and compared to the original. The results of both evaluations lead to a discussion on possible improvements in the metodology that influence the functionality and performance. The resulting improvements are implemented and integrated in RetScan. To further improve performance, a parallelization of RetScan to take advantage of a multi-core architecture or a CUDA-enabled accelerator was designed, coded and evaluated. This evaluation reveals that RetScan achieves its best throughput efficiency using a multi-core architecture only and analysing several images at once. For one image usage, using multi-core only is also the best solution, but with a small speed-up. The usage of CUDA-enabled accelerators is not recom- mended for this scope as the images are small and the cost of the data transfer to and from the accelerator has a severe impact on performance. -

Simulating Linux Clusters on Linux Clusters
Full Circle: Simulating Linux Clusters on Linux Clusters ¡ ¢ ¡ Luis Ceze , Karin Strauss , George Almasi , Patrick J. Bohrer , Jose´ R. Brunheroto , ¡ ¡ ¡ ¡ Calin Cas¸caval , Jose´ G. Castanos˜ , Derek Lieber , Xavier Martorell , ¡ ¡ ¡ Jose´ E. Moreira , Alda Sanomiya , and Eugen Schenfeld £ luisceze,kstrauss ¤ @uiuc.edu £ gheorghe,pbohrer,brunhe,cascaval,castanos,lieber, xavim,jmoreira,sanomiya,eugen ¤ @us.ibm.com ¥ Department of Computer Science, University of Illinois at Urbana-Champaign Urbana, IL 61801 ¦ IBM Thomas J. Watson Research Center Yorktown Heights, NY 10598-0218 § IBM Austin Research Laboratory Austin, TX 78758 Abstract. BGLsim is a complete system simulator for parallel machines. It is currently being used in hardware validation and software development for the BlueGene/L cellular architecture machine. BGLsim is capable of functionally simulating multiple nodes of this machine operating in parallel. It simulates in- struction execution in each node and the communication that happens between nodes. BGLsim allows us to develop, test, and run the exactly same code that will be used in the real system. Using BGLsim, we can gather data that helps us debug and enhance software (including parallel software) and evaluate hard- ware. To illustrate the capabilities of BGLsim, we describe experiments running the NAS Parallel Benchmark IS on a simulated BlueGene/L machine. BGLsim is a parallel application that runs on Linux clusters. It executes fast enough to run complete operating systems and complex MPI codes. 1 Introduction Linux clusters have revolutionized high-performance computing by delivering large amounts of compute cycles at a low price. This has enabled computing at a scale that was previously not affordable to many people.