High-Yielding 18F Radiosynthesis of a Novel Oxytocin Receptor Tracer, a Probe for Nose-To-Brain Oxytocin Uptake in Vivo
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Peptide Synthesis: Chemical Or Enzymatic
Electronic Journal of Biotechnology ISSN: 0717-3458 Vol.10 No.2, Issue of April 15, 2007 © 2007 by Pontificia Universidad Católica de Valparaíso -- Chile Received June 6, 2006 / Accepted November 28, 2006 DOI: 10.2225/vol10-issue2-fulltext-13 REVIEW ARTICLE Peptide synthesis: chemical or enzymatic Fanny Guzmán Instituto de Biología Pontificia Universidad Católica de Valparaíso Avenida Brasil 2950 Valparaíso, Chile Fax: 56 32 212746 E-mail: [email protected] Sonia Barberis Facultad de Química, Bioquímica y Farmacia Universidad Nacional de San Luis Ejército de los Andes 950 (5700) San Luis, Argentina E-mail: [email protected] Andrés Illanes* Escuela de Ingeniería Bioquímica Pontificia Universidad Católica de Valparaíso Avenida Brasil 2147 Fax: 56 32 2273803 E-mail: [email protected] Financial support: This work was done within the framework of Project CYTED IV.22 Industrial Application of Proteolytic Enzymes from Higher Plants. Keywords: enzymatic synthesis, peptides, proteases, solid-phase synthesis. Abbreviations: CD: circular dichroism CLEC: cross linked enzyme crystals DDC: double dimer constructs ESI: electrospray ionization HOBT: hydroxybenzotriazole HPLC: high performance liquid hromatography KCS: kinetically controlled synthesis MALDI: matrix-assisted laser desorption ionization MAP: multiple antigen peptide system MS: mass spectrometry NMR: nuclear magnetic resonance SPS: solution phase synthesis SPPS: solid-phase peptide synthesis t-Boc: tert-butoxycarbonyl TCS: thermodynamically controlled synthesis TFA: trifluoroacetic acid Peptides are molecules of paramount importance in the medium, biocatalyst and substrate engineering, and fields of health care and nutrition. Several technologies recent advances and challenges in the field are analyzed. for their production are now available, among which Even though chemical synthesis is the most mature chemical and enzymatic synthesis are especially technology for peptide synthesis, lack of specificity and relevant. -

Peptide Services090320.Ai
FlexPeptideTM Ensures Delivery & Quality! cGMP Peptide: Up to kilograms can be synthesized Mirror-Image Peptide Drug Discovery Services (Cat.No. SC1211) GenScript constructs libraries with natural L-peptides, which are easily amendable. These libraries are then screened with synthetic mirror-image D-forms of the target proteins to identify potent leads that can guide the synthesis of their mirror-image D-peptides. Competitive Advantages Services Include Delivery Specifications Synthesis and purification of D-enantiomers of target protein Chemically synthesized and purified D-enantiomers of the target protein Synthesis and screening of L-peptide libraries Libraries of chemically synthesized L-peptides Construction and screening of L-peptide phage display libraries Libraries of L-peptide phage display Synthesis of D-enantiomers of isolated peptide leads D-enantiomers of the isolated L-peptide leads Functional characterization data of the leads (if applicable) QC reports Custom Recombinant Peptide Services (Cat.No. SC1082) Due to the fact that long peptides (> 150 residues) or complicated peptides (multiple disulfide bonds) are prohibitively expensive to synthesize chemically. GenScript has developed a proprietary recombinant peptide system that complements our standard chemical peptide synthesis service. The powerful combination of these two-protocol allows GenScript to provide our customers with any peptide of any length on any scale. Competitive Advantages Services Include Key Features • Any length • Superior precision • Any sequence • Outstanding procedure • Any scale • Scalable system Custom Peptide Services • Low cost • Batch-to-Batch consistency FlexPeptideTM Ensures Delivery & Quality! Recommended Purity Levels Standard Peptide Synthesis GenScript proposes a range of different purity levels to help you make the right choice for your application. -

Synthesis of Proteins by Automated Flow Chemistry
Synthesis of Proteins by Automated Flow Chemistry Authors: N. Hartrampf1, A. Saebi1†, M. Poskus1†, Z. P. Gates1, A. J. Callahan1, A. E. Cowfer1, S. Hanna1, S. Antilla1, C. K. Schissel1, A. J. Quartararo1, X. Ye1, A. J. Mijalis1,2, M. D. Simon1, A. Loas1, S. Liu1,3, C. Jessen4, T. E. Nielsen4 and B. L. Pentelute1* 5 Affiliations: 1 Massachusetts Institute of Technology, Department of Chemistry, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. 2 Current address: Harvard Medical School, Department of Genetics, 77 Avenue Louis Pasteur, Boston, 10 MA 02115, USA. 3 Current address: Department of Chemistry, East China Normal University, 3663 North Zhongshan Rd., Shanghai, 200062, China. 4 Novo Nordisk A/S, Novo Nordisk Park, DK-2760 Måløv, Denmark. *Correspondence to: [email protected] 15 † authors contributed equally. Abstract: Ribosomes produce most proteins of living cells in seconds. Here we report highly efficient 20 chemistry matched with an automated fast-flow instrument for the direct manufacturing of peptide chains up to 164 amino acids over 328 consecutive reactions. The machine is rapid - the peptide chain elongation is complete in hours. We demonstrate the utility of this approach by the chemical synthesis of nine different protein chains that represent enzymes, structural units, and regulatory factors. After purification and folding, the synthetic materials display biophysical and enzymatic 25 properties comparable to the biologically expressed proteins. High-fidelity automated flow chemistry is an alternative for producing single-domain proteins without the ribosome. One Sentence Summary: A benchtop automated machine synthesizes protein chains in hours. 30 Main Text: Mechanical pumps, valves, solid supports and computers have transformed the way we perform chemical reactions. -
Peptide Synthesis and Modification As a Versatile Strategy for Probes Construction
University of Pennsylvania ScholarlyCommons Master of Chemical Sciences Capstone Projects Department of Chemistry 8-11-2017 Peptide Synthesis and Modification as a ersatileV Strategy for Probes Construction Jieliang Wang University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/mcs_capstones Part of the Chemistry Commons Wang, Jieliang, "Peptide Synthesis and Modification as a ersatileV Strategy for Probes Construction" (2017). Master of Chemical Sciences Capstone Projects. 7. https://repository.upenn.edu/mcs_capstones/7 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/mcs_capstones/7 For more information, please contact [email protected]. Peptide Synthesis and Modification as a ersatileV Strategy for Probes Construction Abstract Peptide synthesis and modification is a ersatilev chemical biology strategy to construct probes and sensors of a variety of types of biological activity, including protein/protein interactions, protein localization, and proteolysis. In my thesis work, I have made probes for three distinct biological applications. To do so, I have used a combination of solid phase peptide synthesis (SPPS), native chemical ligation (NCL), protein expression, and S-alkylation to construct probes with desired functional groups, while minimizing the perturbation to the native structure. In the first project, I constructed photo- crosslinking probes to study the difference in protein-protein interactions of N-terminal acetylated (N-ac) histone H4 peptide versus non-acetylated histone H4 peptide. One protein was identified yb Western blot with binding preference to N-Ace histone H4 peptide. In the second separate project, I constructed probes to study the toxicity mechanism of proline/arginine dipeptide PRx from amyotrophic lateral sclerosis (ALS) associated gene C9ORF72. -

Custom Peptide Services
Innovative Peptide Solutions Custom Peptide Services Custom & Specialty Peptides Clinical Peptides Peptide Libraries Peptide Pools Peptide Arrays Peptidomimetic & Organic Synthesis Innovative Peptide Solutions JPT’s key technologies are: Custom & Specialty Peptides We are peptide experts with a track record of more than 20 years and offer the largest variety of peptide History chemistries, formats and modifications. JPT Peptide Technologies is a service provider located in Berlin, Germany that has achieved worldwide credi- PepMix™ bility for its commitment to rigorous quality standards Defined antigen spanning peptide pools to and a reputation for developing and implementing stimulate CD4+ and CD8+ T-cells. innovative peptide-based services and research tools for various applications. PepTrack™ Together with its US-subsidiary JPT serves its clientele Peptide libraries of individual peptides offering in the pharmaceutical and biotechnology industries as various specifications and optimization for different well as researchers in universities, governmental and types of assays. non-profit organizations. Clinical Peptides Custom peptides produced for the stringent require- ments of cellular therapy as well as vaccine and Technology & drug development. Application PepStar™ Over the past decade JPT has developed a portfolio of Peptide microarray platform for antibody epitope propietary technologies as well as innovative products dis covery, monitoring of humoral immune responses, and services that have helped to advance the develop- protein-protein interactions and enzyme profiling. ment of new immunotherapies, proteomics and drug discovery. SPOT High-throughput peptide synthesis for T-cell epitope discovery , neo epitope qualification and peptide lead Quality Assurance discovery. JPT is DIN EN ISO 9001:2015 certified and GCLP audited. SpikeTides™ Light and stable isotope-labeled or quantified peptides for mass spectrometry based proteomics assays. -

Synthesis, Characterization and Evaluation of Peptide Nanostructures for Biomedical Applications
molecules Review Synthesis, Characterization and Evaluation of Peptide Nanostructures for Biomedical Applications Fanny d’Orlyé, Laura Trapiella-Alfonso , Camille Lescot, Marie Pinvidic, Bich-Thuy Doan and Anne Varenne * Chimie ParisTech PSL, CNRS 8060, Institute of Chemistry for Life and Health (i-CLeHS), 75005 Paris, France; [email protected] (F.d.); [email protected] (L.T.-A.); [email protected] (C.L.); [email protected] (M.P.); [email protected] (B.-T.D.) * Correspondence: [email protected]; Tel.: +33-1-8578-4252 Abstract: There is a challenging need for the development of new alternative nanostructures that can allow the coupling and/or encapsulation of therapeutic/diagnostic molecules while reducing their toxicity and improving their circulation and in-vivo targeting. Among the new materials using natural building blocks, peptides have attracted significant interest because of their simple structure, relative chemical and physical stability, diversity of sequences and forms, their easy functionalization with (bio)molecules and the possibility of synthesizing them in large quantities. A number of them have the ability to self-assemble into nanotubes, -spheres, -vesicles or -rods under mild conditions, which opens up new applications in biology and nanomedicine due to their intrinsic biocompatibility and biodegradability as well as their surface chemical reactivity via amino- and carboxyl groups. In order to obtain nanostructures suitable for biomedical applications, the structure, size, shape and surface chemistry of these nanoplatforms must be optimized. These properties depend directly on Citation: d’Orlyé, F.; the nature and sequence of the amino acids that constitute them. -

And Light-Directed Peptide Microarray Synthesis
Development of Chip-Based Electrochemically- and Light-Directed Peptide Microarray Synthesis by Pallav Kumar A Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy Approved October 2013 by the Graduate Supervisory Committee: Neal Woodbury, Chair James Allen Stephen Johnston ARIZONA STATE UNIVERSITY December 2013 ABSTRACT Peptide microarrays may prove to be a powerful tool for proteomics research and clinical diagnosis applications. Fodor et al. and Maurer et al. have shown proof-of- concept methods of light- and electrochemically-directed peptide microarray fabrication on glass and semiconductor microchips respectively. In this work, peptide microarray fabrication based on the abovementioned techniques were optimized. In addition, MALDI mass spectrometry based peptide synthesis characterization on semiconductor microchips was developed and novel applications of a CombiMatrix (CBMX) platform for electrochemically controlled synthesis were explored. We have investigated performance of 2-(2-nitrophenyl)propoxycarbonyl (NPPOC) derivatives as photo-labile protecting group. Specifically, influence of substituents on 4 and 5 positions of phenyl ring of NPPOC group on the rate of photolysis and the yield of the amine was investigated. The results indicated that substituents capable of forming a π-network with the nitro group enhanced the rate of photolysis and yield. Once such properly substituted NPPOC groups were used, the rate of photolysis/yield depended on the nature of protected amino group indicating that a different chemical step during the photocleavage process became the rate limiting step. We also focused on electrochemically-directed parallel synthesis of high-density peptide microarrays using the CBMX technology referred to above which uses electrochemically generated acids to perform patterned chemistry. -

Repurposing Modular Polyketide Synthases and Non-Ribosomal Peptide Synthetases for Novel Chemical Biosynthesis
Downloaded from orbit.dtu.dk on: Oct 05, 2021 Repurposing Modular Polyketide Synthases and Non-ribosomal Peptide Synthetases for Novel Chemical Biosynthesis Hwang, Soonkyu; Lee, Namil; Cho, Suhyung; Palsson, Bernhard; Cho, Byung-Kwan Published in: Frontiers in Molecular Biosciences Link to article, DOI: 10.3389/fmolb.2020.00087 Publication date: 2020 Document Version Publisher's PDF, also known as Version of record Link back to DTU Orbit Citation (APA): Hwang, S., Lee, N., Cho, S., Palsson, B., & Cho, B-K. (2020). Repurposing Modular Polyketide Synthases and Non-ribosomal Peptide Synthetases for Novel Chemical Biosynthesis. Frontiers in Molecular Biosciences, 7, [87]. https://doi.org/10.3389/fmolb.2020.00087 General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Users may download and print one copy of any publication from the public portal for the purpose of private study or research. You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. fmolb-07-00087 May 13, 2020 Time: 17:31 # 1 REVIEW published: 15 May -

Solid Phase Peptide Synthesis (SPPS), Strategies, Resins and Comparison with Fmoc-Strategy General Scheme of SPPS
Solid phase peptide synthesis (SPPS), strategies, resins and comparison with Fmoc-strategy General scheme of SPPS attach to linker deprotect amino function couple n times deprotection and coupling cleave N-α-protecting groups • two mainly used N-α-protecting groups Fmoc Boc Boc-protecting group Boc = tert. Butyloxycarbonyl or tert. Butoxycarbonyl stable to bases and nucleophiles unaffected by catalytic hydrogenation deprotection with TFA is rapid Introduction of the Boc-group Di-tert-butyl-dicarbonate 2-(tert. Butoxycarbonyl- = Boc-anhydride = (t-boc)2O oxyimino)-2-phenylacetonitril = Boc-NO O O O O O O O O N N • both commercially available • storage in refrigerator for extended periods •(t-boc)2O is more expensive • preparation: „Kates S. A., Albericio F. (ed): Solid-Phase Synthesis, A practical guide, Marcel Dekker Inc. 2000, p. 105-107“ N-α-Boc protected amino acids •alreadyN-α-Boc protected amino acids can simply be bought from firms like „Novabiochem“ O e.g. Boc-Ala-OH O N COOH H Cleavage of the Boc-group TFA E1-elimination CO2 cleavage TFA is volatile and can be easily removed in vacuum! Resins for Boc SPPS Resins for preparing peptide acids Merrifield (Chloromethylstyrene-divinylbenzene) - was the standard support for the synthesis of peptide acids by Boc SPPS - now only used in the synthesis of small to medium sized peptides, because the benzylic ester resin linkage is not completely stable towards repetitive treatment with TFA O H Cl , DMF N boc O - + + BocAS Cs ,KI R - attachment of the C-terminal residue is achieved by heating -

Synthesis of Peptide Radiopharmaceuticals for the Therapy and Diagnosis of Tumor Diseases
Molecules 2013, 18, 3379-3409; doi:10.3390/molecules18033379 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Synthesis of Peptide Radiopharmaceuticals for the Therapy and Diagnosis of Tumor Diseases Mazen Jamous, Uwe Haberkorn and Walter Mier * Department of Nuclear Medicine, University Hospital Heidelberg, Im Neuenheimer Feld 400, D-69120 Heidelberg, Germany * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +49-6221-56-7720; Fax: +49-6221-65-33629. Received: 29 December 2012; in revised form: 25 February 2013 / Accepted: 7 March 2013 / Published: 14 March 2013 Abstract: Despite the advances in molecular biology and biochemistry, the prognosis of patients suffering from tumor diseases remains poor. The limited therapeutic success can be explained by the insufficient performance of the common chemotherapeutic drugs that lack the ability to specifically target tumor tissues. Recently peptide radiopharmaceuticals have been developed that enable the concurrent imaging and therapy of tumors expressing a specific target. Here, with a special emphasis on the synthesis of the building blocks required for the complexation of metallic radioisotopes, the requirements to the design and synthesis of radiolabeled peptides for clinical applications are described. Keywords: radionuclides; chelator; prosthetic groups; carrier molecules; peptides; medicinal application; radiopharmaceutical; diagnostic imaging; radiotherapeutics 1. Introduction The incidence of human malignant tumor diseases is still increasing worldwide. Generally, cancer treatment can be performed using one or a combination of the following methods: surgery, chemotherapy and radiation therapy. Their side effects limit the efficiency of chemo- and radiotherapeutic agents, but can be avoided and a much more effective therapy is possible if the drugs used have tumor selectivity. -
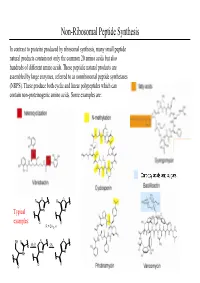
Non-Ribosomal Peptide Synthesis
Non-Ribosomal Peptide Synthesis In contrast to proteins produced by ribosomal synthesis, many small peptide natural products contain not only the common 20 amino acids but also hundreds of different amino acids. These peptidic natural products are assembled by large enzymes, referred to as nonribosomal peptide synthetases (NRPS). These produce both cyclic and linear polypeptides which can contain non-proteinogenic amino acids. Some examples are: O R R O Typical N N examples: O O R = CH3, H SH O S S -H2OOx. NH N N O O O Non-Ribosomal Peptide Synthesis: Initiation (A) Non-ribosomal peptide bio-synthesis requires A proper assembly and modification of a large multienzyme complexes including the addition of the prosthetic moiety 4'-phosphopantetheinyl cofactor (ppan) to a conserved serine residue of chain of peptidyl carrier protein (PCP). (B) The first step of peptide assembly is similar to ribosomal protein synthesis: an amino acid is B activated by trans-esterification with ATP to afford the corresponding aminoacyl-adenylate. The domains A1 and A2 each bind to a specific amino acid through non-covalent interactions and catalyze the trans-esterification to produce each aminoacyl- adenylate. “A” = adenylation domain. (C) Domain A also catalyzes the transfer of each C aminoacyl-adenylate onto the free thiol group of PCP- ppan within its module. This establishes a covalent linkage between enzyme and substrate. At this stage the substrate can undergo modifications such as epimerization or N-methylation by neighboring editing or “E” domains. Non-Ribosomal Peptide Synthesis: Elongation (D) Assembly of the product occurs by a series of D peptide bond formation steps (elongation) between the downstream building block with its free amine and the carboxy-thioester of the upstream substrate. -

Custom Peptide Synthesis Faqs
Biomatik Tel: (519) 489-7195, (800) 836-8089 Fax: (519) 231-0140, (877) 221-3515 Email: [email protected] http://www.biomatik.com Peptide Synthesis FAQs Version 4.1, Revision 2019-10-06 Below are some frequently asked questions about the Biomatik Peptide Synthesis service. If you have any other questions, please contact us at [email protected] and we will be happy to assist you. 1. Do you have customer testimonials and publications citing your peptide synthesis service? Biomatik has been trusted among top scientists and researchers from around the world for quality products and custom services since 2002. To date, Biomatik has delivered 70,000+ custom made products to our customers worldwide. Learn why Biomatik has been trusted by thousands of researchers worldwide by viewing our customer testimonials, selected publications and 3,000+ publications citing our fine products and services at Google Scholar. 2. What is the typical turn-around time for peptide synthesis? How do you ship and how long does it take to ship to me? Our typical turn-around time is 2-3 weeks for a standard purified peptide under 30 amino acids. The turn-around time varies depending on the peptide length, solubility and difficulty. All peptides will be shipped in lyophilized powder, at room temperature. It is overnight delivery within USA/Canada, and takes typically 3-4 days to reach researchers in other countries. 3. Which analytical data do you provide for peptides? Each peptide comes with CoA, MS spectra, and HPLC chromatograms. See Sample QA Report. We can provide additional analysis if required, e.g.