1 1 New Insights Into the Composition of Indian Yellow and Its Use in A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Museum of Economic Botany, Kew. Specimens Distributed 1901 - 1990
Museum of Economic Botany, Kew. Specimens distributed 1901 - 1990 Page 1 - https://biodiversitylibrary.org/page/57407494 15 July 1901 Dr T Johnson FLS, Science and Art Museum, Dublin Two cases containing the following:- Ackd 20.7.01 1. Wood of Chloroxylon swietenia, Godaveri (2 pieces) Paris Exibition 1900 2. Wood of Chloroxylon swietenia, Godaveri (2 pieces) Paris Exibition 1900 3. Wood of Melia indica, Anantapur, Paris Exhibition 1900 4. Wood of Anogeissus acuminata, Ganjam, Paris Exhibition 1900 5. Wood of Xylia dolabriformis, Godaveri, Paris Exhibition 1900 6. Wood of Pterocarpus Marsupium, Kistna, Paris Exhibition 1900 7. Wood of Lagerstremia parviflora, Godaveri, Paris Exhibition 1900 8. Wood of Anogeissus latifolia , Godaveri, Paris Exhibition 1900 9. Wood of Gyrocarpus jacquini, Kistna, Paris Exhibition 1900 10. Wood of Acrocarpus fraxinifolium, Nilgiris, Paris Exhibition 1900 11. Wood of Ulmus integrifolia, Nilgiris, Paris Exhibition 1900 12. Wood of Phyllanthus emblica, Assam, Paris Exhibition 1900 13. Wood of Adina cordifolia, Godaveri, Paris Exhibition 1900 14. Wood of Melia indica, Anantapur, Paris Exhibition 1900 15. Wood of Cedrela toona, Nilgiris, Paris Exhibition 1900 16. Wood of Premna bengalensis, Assam, Paris Exhibition 1900 17. Wood of Artocarpus chaplasha, Assam, Paris Exhibition 1900 18. Wood of Artocarpus integrifolia, Nilgiris, Paris Exhibition 1900 19. Wood of Ulmus wallichiana, N. India, Paris Exhibition 1900 20. Wood of Diospyros kurzii , India, Paris Exhibition 1900 21. Wood of Hardwickia binata, Kistna, Paris Exhibition 1900 22. Flowers of Heterotheca inuloides, Mexico, Paris Exhibition 1900 23. Leaves of Datura Stramonium, Paris Exhibition 1900 24. Plant of Mentha viridis, Paris Exhibition 1900 25. Plant of Monsonia ovata, S. -

Personal Enrichment Courses SUPPLY LIST
Personal Enrichment Courses SUPPLY LIST Beginning Acrylics Intermediate Acrylics Instructor: Patti Overholt Instructor: Patti Overholt Niceville Campus Niceville Campus Please try to purchase Galeria Acrylic Paints Supply List (Windsor Newton) for best color mixing results. 1. CANVAS: One 8x10 Canvas Panel Supply List One 9 x 12 Canvas Panel 1. CANVAS: One 8x10 Gallery Wrapped Canvas Three 8x10 Canvas Panels 2. BRUSHES: 2. BRUSHES: #1 inch and a #0.5 inch Flat Brush #1 inch and a #0.5 inch Flat Brush #4 inch and a #8 Filbert #4 inch and a #8 Filbert #8 inch Round Brush #8 inch Round Brush A fan Brush A fan Brush A one inch craft brush A one inch craft brush 3 Palettes Knives, Small, Med. and Large 3. ACRYLIC PAINT: 3. ACRYLIC PAINT: (Starter Sets are available online and at local craft stores. Hobby (Starter Sets are available online and at local craft stores. Hobby Lobby has the best coupon offers. PLEASE avoid cheap paints as Lobby has the best coupon offers. PLEASE avoid cheap paints as colors are off and the pigments are thin.) colors are off and the pigments are thin.) • Ultramarine Blue • Ultramarine Blue • Cerulean Blue • Cerulean Blue • Alizarin Crimson • Alizarin Crimson • Rose Pink • Rose Pink • Cadmium Red Medium • Cadmium Red Medium • Cadmium Yellow Medium • Cadmium Yellow Medium • Yellow Ochre • Yellow Ochre • Indian Yellow • Indian Yellow • Titanium White • Titanium White • Unbleached Titanium (Buff • Unbleached Titanium (Buff White) White) • Burnt Umber • Burnt Umber • Acrylic Extender • Acrylic Extender MISCELLANEOUS: MISCELLANEOUS: Brush Holder for water Plastic Palette Plastic Bottle with water Styrofoam Trays Paper Towels Small jar Golden Moulding Paste Package of Handy or Baby Wipes Brush Holder for water Plastic Bottle with water Paper Towels Package of Handy or Baby Wipes Saran Wrap Personal Enrichment Courses SUPPLY LIST Acrylic Painting Have Fun Drawing Instructor: Marvin Tweedy Instructor: Patti Overholt DeFuniak Springs Campus Niceville Campus Supply List Supply List 1. -

The ISCC-NBS Method of Designating Colors and a Dictionary of Color Names
Uc 8 , .Department of Commerce Na Canal Bureau of Standards Circular UNITED STATES DEPARTMENT OF COMMERCE • Sinclair Weeks, Secretary NATIONAL BUREAU OF STANDARDS • A. V. Astin, Director The ISCC-NBS Method of Designating Colors and a Dictionary of Color Names National Bureau of Standards Circular 553 Issued November 1, 1955 For sale by the Superintendent of Documents, U. S. Government Printing Office, Washington 25, D. C. Price 32 7 1 National Bureau of Standards NOV 1 1955 8 (0*118 QC 00 U555 Cop. 1 Preface I^Ever since the language of man began to develop, words or expressions have been used first to indicate and then to describe colors. Some of these have per- sisted throughout the centuries and are those which refer to the simple colors or ranges such as red or yellow. As the language developed, more and more color names were invented to describe the colors used by art and industry and in late years in the rapidly expanding field of sales promotion. Some of these refer to the pigment or dye used, as Ochre Red or Cochineal, or a geographical location of its source such as Naples Yellow or Byzantium. Later when it became clear that most colors are bought by or for women, many color names indicative of the beauties and wiles of the fan- sex were introduced, as French Nude, Heart’s Desire, Intimate Mood, or Vamp. Fanciful color names came into vogue such as Dream Fluff, Happy Day, Pearly Gates, and Wafted Feather. Do not suppose that these names are without economic importance for a dark reddish gray hat for Milady might be a best seller ; if advertised as Mauve Wine whereas it probably would not if the color were called Paris Mud. -

Gamblin Provides Is the Desire to Help Painters Choose the Materials That Best Support Their Own Artistic Intentions
AUGUST 2008 Mineral and Modern Pigments: Painters' Access to Color At the heart of all of the technical information that Gamblin provides is the desire to help painters choose the materials that best support their own artistic intentions. After all, when a painting is complete, all of the intention, thought, and feeling that went into creating the work exist solely in the materials. This issue of Studio Notes looks at Gamblin's organization of their color palette and the division of mineral and modern colors. This visual division of mineral and modern colors is unique in the art material industry, and it gives painters an insight into the makeup of pigments from which these colors are derived, as well as some practical information to help painters create their own personal color palettes. So, without further ado, let's take a look at the Gamblin Artists Grade Color Chart: The Mineral side of the color chart includes those colors made from inorganic pigments from earth and metals. These include earth colors such as Burnt Sienna and Yellow Ochre, as well as those metal-based colors such as Cadmium Yellows and Reds and Cobalt Blue, Green, and Violet. The Modern side of the color chart is comprised of colors made from modern "organic" pigments, which have a molecular structure based on carbon. These include the "tongue- twisting" color names like Quinacridone, Phthalocyanine, and Dioxazine. These two groups of colors have unique mixing characteristics, so this organization helps painters choose an appropriate palette for their artistic intentions. Eras of Pigment History This organization of the Gamblin chart can be broken down a bit further by giving it some historical perspective based on the three main eras of pigment history – Classical, Impressionist, and Modern. -

MODERN WATERCOLOR III Fair FR Fair G Granulating J Semi-Opaque NA Not Yet Tested H Opaque
ASTM LF GOLDEN LF STAIN OPACITY I Excellent EX Excellent Semi-Staining k Semi-Transparent II Very Good GD Good Staining i Transparent MODERN WATERCOLOR III Fair FR Fair G Granulating j Semi-Opaque NA Not Yet Tested h Opaque Item # Series Color Color Index Name Pigment Name ASTM Opacity Granulating Staining 1-4 Light-fastness 7000525 1 Ardoise Gray PBk19 Hydrated Aluminum Silicate I i G 7000135 1 Aureolin Modern PW6, PY150 Titanium Dioxide Rutile, Nickel Complex Azo I j 7000126 4 Benzimidazolone Yellow PY154 Benzimidazolone Yellow H3G I k 7000115 3 Bismuth Vanadate Yellow PY184 Bismuth Vanadate EX k 7000415 2 Bohemian Green Earth PBk7, PG7, PR101, PY42 Nearly Pure Amorphous Carbon, Chlorinated Copper I k G Phthalocyanine, Synthetic Iron Oxide, Synthetic Iron Oxide 7000470 1 Burnt Sienna (Natural) Pbr7 Calcined Natural Iron Oxide I j G 7000480 1 Burnt Umber (Natural) Pbr7 Calcined Natural Iron Oxide containing Manganese I j G 7000180 4 Cadmium Orange PO20 Cadmium (Sulfo-Selenide) - (CC) I j 7000220 4 Cadmium Red Deep PR108 Cadmium (Sulfo-Selenide) - (CC) I j G 7000195 4 Cadmium Red Light PR108 Cadmium (Sulfo-Selenide) - (CC) I j G 7000215 4 Cadmium Red Medium PR108 Cadmium (Sulfo-Selenide) - (CC) I j 7000170 4 Cadmium Yellow Deep PY35 Cadmium Zinc Sulfide - (CC) I k 7000120 4 Cadmium Yellow Light PY35 Cadmium Zinc Sulfide - (CC) I k 7000130 4 Cadmium Yellow Medium PY35 Cadmium Zinc Sulfide - (CC) I k 7000105 4 Cadmium Yellow Primrose PY35 Cadmium Zinc Sulfide - (CC) I k 7000500 1 Carbon Black PBk7 Nearly Pure Amorphous Carbon I h 7000330 -

Brochure Old Holland Classic Oil Colours Drying Time & Transparency
Old Holland Classic Oil Colours Drying time & Transparency Transparency: * = Transparent **** = Opaque Drying time: F = Fast, M = Medium, S = Slow Oil content: H = High, M = Medium, L = Low, VL = Very Low Colour name Colour index Pigment classification Trans- light- Drying Oil parency fastness time content A1 Titanium white PW6 Titanium dioxide **** 7/8 m l A2 Zinc white PW4 Zinc oxide *** 7/8 m l 3 Lead white PW1 Basic lead carbonate **** 7/8 f vl 4 Flake white no.1 PW1-PW4 Basic lead carbonate zinc oxide **** 7/8 m vl A5 Mixed white no.2 PW6-PW4 Titanium dioxide- zinc oxide **** 7/8 m l A6 Old Holland yellow PW4-PW6- Zinc oxide- Titanium dioxide- Natural ochre **** 7/8 m l light PY43 B8 Old Holland yellow PW4-PW6-PY- Zinc oxide-titanium dioxide-mono.azo-Natural ochre **** 7/8 m l deep 74-PY43 B7 Old Holland yellow PW4-PW6- Zinc oxide-titanium dioxide-monoazo-Natural ochre *** 7/8 m l medium PY74-PY43 B103 Brilliant yellow PW4-PW6- Zinc oxide- Titanium dioxide- Disazo- Diketo pyrollo *** 7/8 m l light PY83-PO73 pyrrole B106 Brilliant yellow PW4-PW6- Zinc oxide- Titanium dioxide- Nickel titanate- Diketo- **** 7/8 m l PY53-PO73 Pyrrolo- Pyrolle B109 Brilliant yellow- PW4-PW6- Zinc white- Titanium dioxide- Nickel titanate- Diketo- **** 7/8 m l reddish PY53-PO73 Pyrrolo- Pyrolle B112 Napels yellow PW4-PW6- Zinc oxide- Titanium dioxide- Nickel titanate- Disazo **** 7/8 m l reddish extra PY53-PO43 B115 Flesh tint PW4-PW6- Zinc oxide- Titanium dioxide- Zinc iron oxide- *** 7/8 m l PY119-PR2 Condensed azo B118 Indian yellow- PY95-PY129 Azo condensation- -

Syllabus 29002 ART 315.001 Color, S20, 5:30 – 8:10 Pm, MW, School of Art Graphic Design B121 Professor Peter Andrew, [email protected], 936-468-4804 Ext
Syllabus 29002 ART 315.001 Color, S20, 5:30 – 8:10 pm, MW, School of Art graphic design B121 Professor Peter Andrew, [email protected], 936-468-4804 ext. 4451, office B127, advising/office hours by appointment. Students registered in this class are responsible for fulfilling the requirements in this syllabus in order to earn a passing grade. Description: ART 315 Color - 3 semester hours, 6 hours studio, 6 hours independent study per week. Practice, theory, and study of color in art, media, and design. Levels A, B. Prerequisite: ART 110. This course is the study and practice of color in design. Students research, study, and mix color in a variety of projects. Projects are color assignments and free studies aimed at enabling a person to see, control, and eventually predict or master color effect. The instructor and visiting artists will demonstrate techniques and materials. The midterm and final exam presentations require you to prepare and conduct a live color demonstration, in addition to an exhibition of your personal color studies. Intended Course Learning Outcomes • Students demonstrate proficiency in art studio practices, to apply in a major studio focus area. • Students exhibit competency applying technical and problem solving skills appropriate to their art studio focus. • Students develop visual skills and creativities. • Students compare self-progress to models of excellence in art with in and outside of class activities. • Students actively invent, produce and showcase their art skills to the public in art exhibits. Student Learning Outcomes • Students demonstrate abilities to explore creative options within clearly defined limits. • Students demonstrate abilities to follow directions and to complete projects on-time building craftsmanship. -

Color Chart Includes Those Colors Made from Inorganic Pigments, That Is, Metal Ores Dug from the Earth
GAMBLIN ARTISTS COLORS GAMBLIN ARTISTS OIL COLORS Artists Oil mineral inorganic colors modern organic colors Colors • All colors made from metals (Cadmium, Cobalt, Iron, etc.) are “inorganic” • Carbon based pigments are “organic” • 19th century colors of the Impressionists and the colors of Classical and Renaissance era painters • 20th century colors • High pigment load, low oil absorption • Most pigments available in a warm and cool version (ex. Phthalo Green, Phthalo Emerald) • Colors easily grey-down in mixtures, excellent for painting natural colors and light • Best choice for high key painting, bright tints • Mostly opaque with a few semi-transparent and transparent colors • Mostly transparent, with some semi-transparent colors Impressionist 20th Century CADMIUM CHARTREUSE CADMIUM LEMON CADMIUM YELLOW LIGHT CADMIUM YELLOW MEDIUM CADMIUM YELLOW DEEP HANSA YELLOW LIGHT HANSA YELLOW MEDIUM HANSA YELLOW DEEP INDIAN YELLOW CADMIUM ORANGE CADMIUM ORANGE DEEP CADMIUM RED LIGHT CADMIUM RED MEDIUM CADMIUM RED DEEP PERMANENT ORANGE TrANSPARENT OrANGE NAPTHOL RED NAPTHOL SCARLET PERYLENE RED white · grey · black ALIZARIN CrIMSON MANGANESE VIOLET COBALT VIOLET ULTRAMARINE VIOLET ALIZARIN PERMANENT QUINACRIDONE RED QUINACRIDONE MAGENTA QUINACRIDONE VIOLET DIOXAZINE PURPLE TITANIUM WHITE RADIANT WHITE TITANIUM ZINC WHITE QUICK DRY WHITE FLAKE WHITE REPLACEMENT ULTRAMARINE BLUE COBALT BLUE PrUSSIAN BLUE CERULEAN BLUE COBALT TEAL INDANTHRONE BLUE PHTHALO BLUE CERULEAN BLUE HUE MANGANESE BLUE HUE PHTHALO TURQUOISE FASTMATTE TITANIUM WHITE ZINC WHITE -
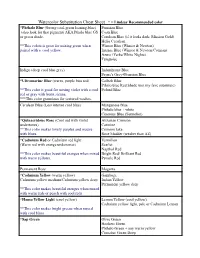
Watercolor Substitution Cheat Sheet * = Lindsay Recommended Color
Watercolor Substitution Cheat Sheet * = Lindsay Recommended color *Phthalo Blue (Strong cool-green leaning-blue) Prussian Blue (also look for that pigment) AKA Pthalo blue GS Cyan Blue or green shade. Cerulean Blue (if it looks dark: Mission Gold) Helio Cerulean **This colors is great for mixing green when Winsor Blue (Winsor & Newton) paired with a cool yellow. Intense Blue (Winsor & Newton/Cotman) Azure (Yarka/White Nights) Turquoise Indigo (deep cool blue grey) Indanthrone Blue Payne's Grey+Prussian Blue *Ultramarine Blue (warm, purple bias red) Colbalt Blue Pthalo blue Red Shade (not my fave substitute) **This color is good for mixing violet with a cool Poland Blue red or gray with burnt sienna. ***This color granulates for textured washes. Cerulean Blue (Less intense cool blue) Manganese Blue Phthalo blue + white Cinerous Blue (Sennelier) *Quinacridone Rose (Cool red with violet Alizarian Crimson undertones) Carmine **This color makes lovely purples and mauve Crimson lake with blues. Rose Madder (weaker than AZ) *Cadmium Red or Cadmium red light Vermilion (Warm red with orange undertones) Scarlet Napthol Red **This color makes beautiful oranges when mixed Bright Red/ Brilliant Red with warm yellows. Pyrrole Red Permanent Rose Magenta *Cadmium Yellow (warm yellow) Gamboge Cadmium yellow medium/Cadmium yellow deep Indian Yellow Permanent yellow deep **This color makes beautiful oranges when mixed with warm reds or peach with cool reds *Hansa Yellow Light (cool yellow) Lemon Yellow (cool yellow) Cadmium yellow light, pale or Cadmium -

Colour Chart.Indd
The Paints, Past and Present Details and Descriptions of Colours This section gives assessments for certain determinable characteristics of all the colours presently in my range, as well as a few informal remarks on their qualities and idiosynchrasies when used. Following the pattern already laid out on all my tube and can labels, and on my more recent colour charts, I give: (1) The Colour Index Number: This is an international system for classifying and identifying pigments solely by their (often very complex) chemical formulae( e.g P(igment) R(ed) 106, Mercuric Sulphide known as Genuine Vermilion). The use of vague traditional or invented colour names is thus clarifi ed, and so, in theory at least, is the vexed matter of what pigments manufacturers actually put into their paints. If a colourman is honest, each constituent pigment in a paint can be specifi ed precisely, and the practice of secretly adulterating or even completely substituting cheaper alternatives is made impossible. Assuming, that is, the colourman is honest…I can certainly state that there are no secret additions to any of the paints in my range. What you read as the C.I number on the label is what you get. (2) Estimated Relative Drying Speed: Bearing in mind that all drying speeds will be affected by temperature, humidity and light levels, these give a broad calibration of comparative speeds, from the Very Fast, such as the Umbers, many of which, if used neat, will be touch dry within a hot summer day, to the Very Slow, which in unmixed state, might take up to a week. -
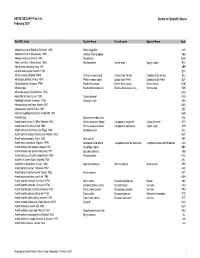
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

A Visit to North Spain at the Time of the Eclipse of 1860
422 VACATION TOURISTS, AND [Spain and Eclipse. 12. A VISIT TO NORTH SPAIN AT THE TIME OF THE ECLIPSE. BY EKANCIS GALTON, F.RS. A direction was given to my summer rambles by the desire of witnessing the solar eclipse of last June, and by the fact that the path of its totality, where nearest to England, lay across a country which I ardently longed to visit. The result was, that I applied for permission, and obtained it, to form one of the party of astronomers who, under the leadership of the Astronomer-Eoyal, were taken by H.M.S. Himalaya to Spain. The Himalaya is truly a noble vessel, and we were right imperially treated. Those whose experience has been drawn from coasting passenger-steamers, in English or Mediterranean waters, would hardly credit that anything floated comparable in spaciousness and luxury to this magnificent ship. And she is as fast and as easy, excepting a tendency to roll, as she is spacious and comfortable ; for we steamed out of Plymouth Sound on a Saturday forenoon, so steadily, that I hardly knew we were moving ; and on the Sunday night we were going at half-power, because we were too near the Spanish coast, whose bold outlines lay in full view on the early Monday morning. It was therefore with one of those feelings of contrast so often enjoyed by travellers, that I, with my eyes still toned to that dim English daylight in which we had just bade fare- well to our shores, found myself paddling up the Bilbao river in a small shore-going craft, under a full flood of southern F.