An Outline for Professionals Their Impact from Relatively Minor Disability (Eg
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Goltz Syndrome Nsu-Com / Larkin Community Hospital Presenters: Ann R Eed, Do, Hyunhee Park, Do, Julie Frederickson , Do
GOLTZ SYNDROME NSU-COM / LARKIN COMMUNITY HOSPITAL PRESENTERS: ANN R EED, DO, HYUNHEE PARK, DO, JULIE FREDERICKSON , DO. PROGRAM DIRECTOR: STANLEY SKOPIT, DO, MSE, FAOCD Case presentation 17 year old female with established diagnosis of Goltz syndrome presented to our office Jan. 2011 with c/o “Dry skin and itchy scalp” PE: Syndromic facies w/ aniridia, microphthalmia, short stature, sparse hair, hypodontia, syndactyly, blaschko linear hyper and hypopigmentation, perioral papillomas, scaly scalp and xerotic skin Dx: Xerosis Cutis , Seborrhea and alopecia in patient with Goltz Tx: Ketoconazole 2% shampoo MWF alt with T/Sal Lidex solution BID x 2 weeks to scalp Cerave/Cetaphil to body Biotin 2500 mcg daily Bx’s: 3/8/11 Shave biopsy (R labial commissure) - Verruca with candidiasis 3/22/11 Shave biopsy (L labial commissure) - Impetiginized Verruca with candidiasis Ketoconazole 2% cream BID given for topical treatment Goltz Syndrome Overview Focal Dermal Hypoplasia or Goltz-Gorlin syndrome Rare genodermatosis Multiple abnormalities of mesodermal and ectodermal tissues First described by Dr. Goltz in 1962 Approximately 300 reported cases worldwide Inheritance X-linked dominant 90% female Lethal in males with non-mosaic hemizygous mutations 10% affected individuals: males with genomic or functional mosaicism 95% of cases are sporadic Gene locus Xp11.23 Mutation in PORCN gene lack of Wnt signaling Variability in clinical severity (lyonization) Goltz Syndrome Cutaneous Findings Wu M-C et al. / Dermatologica Sinica 29 (2011) 59-62 Wang -

Blueprint Genetics Ectodermal Dysplasia Panel
Ectodermal Dysplasia Panel Test code: DE0401 Is a 25 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of ectodermal dysplasia (hidrotic or hypohidrotic) or Ellis-van Creveld syndrome. About Ectodermal Dysplasia Ectodermal Dysplasia (ED) is a group of closely related conditions of which more than 150 different syndromes have been identified. EDs affects the development or function of teeth, hair, nails and sweat glands. ED may present as isolated or as part of a syndromic disease and is commonly subtyped according to sweating ability. The clinical features of the X-linked and autosomal forms of hypohidrotic ectodermal dysplasia (HED) can be indistinguishable and many of the involved genes may lead to phenotypically distinct outcomes depending on number of defective alleles. The most common EDs are hypohidrotic ED and hydrotic ED. X-linked hypohidrotic ectodermal dysplasia (HED) is caused by EDA mutations and explain 75%-95% of familial HED and 50% of sporadic cases. HED is characterized by three cardinal features: hypotrichosis (sparse, slow-growing hair and sparse/missing eyebrows), reduced sweating and hypodontia (absence or small teeth). Reduced sweating poses risk for episodes of hyperthermia. Female carriers may have some degree of hypodontia and mild hypotrichosis. Isolated dental phenotypes have also been described. Mutations in WNT10A have been reported in up to 9% of individuals with HED and in 25% of individuals with HED who do not have defective EDA. Approximately 50% of individuals with heterozygous WNT10A mutation have HED and the most consistent clinical feature is severe oligodontia of permanent teeth. -

Goltz Syndrome) Head and Neck Surgery Manila Central University – Filemon D
CASE REPORTS PHILI pp INE JOURNAL OF OTOLARYNGOLOGY -HEAD AND NECK SURGERY VOL . 32 NO. 2 JULY – DECEMBER 2017 John Emmanuel L. Ong, MD1 Unilateral Tonsilar Hypertrophy Emmanuel Tadeus S. Cruz, MD1,2 Clydine Maria Antonette G. Barrientos, MD1,3 in a 4-Year-Old Girl with Focal Dermal Hypoplasia 1 Department of Otorhinolaryngology (Goltz Syndrome) Head and Neck Surgery Manila Central University – Filemon D. Tanchoco Medical Foundation Hospital 2 Department of Otorhinolaryngology Head and Neck Surgery Quezon City General Hospital ABSTRACT 3 Department of Otorhinolaryngology Objective: To report a case of unilateral tonsillar hypertrophy resulting in severe Obstructive Sleep Head and Neck Surgery Makati Medical Center Apnea in a 4-year-old girl with focal dermal hypoplasia (FDH, Goltz or Goltz-Gorlin) Syndrome. Methods: Design: Case Report Setting: Tertiary Private Teaching Hospital Patient: One Correspondence: Dr. Emmanuel Tadeus S. Cruz Results: A 4-year-old girl with Goltz Syndrome (classical features of cutaneous and osteopathic Department of Otorhinolaryngology – Head and Neck Surgery Manila Central University – Filemon D. Tanchoco Medical disorders since birth) and unilateral tonsillar hypertrophy manifested with snoring and apneic Foundation Hospital Epifanio de los Santos Ave., Caloocan City 1400 episodes at two years of age. Polysomnography revealed severe Obstructive Sleep Apnea Philippines and Arterial Blood Gases revealed metabolic acidosis with hypoxemia. A tonsillectomy and Phone: (632) 367 2031 loc 1212 Email: [email protected] -

Ectodermal Dysplasia (Generic Term)
Ectodermal Dysplasia (generic term) Authors: Doctor Kathleen Mortier1, Professor Georges Wackens1 Creation date: September 2004 Scientific Editor: Professor Antonella Tosti 1Department of stomatology and maxillofacial surgery, AZ VUB Brussels, Belgium [email protected] Definition Clinical classification of Ectodermal dysplasias References Definition Ectodermal dysplasias (EDs) are a heterogeneous group of disorders characterized by developmental dystrophies of ectodermal structures, such as hypohidrosis, hypotrichosis, onychodysplasia and hypodontia or anodontia. About 160 clinically and genetically distinct hereditery ectodermal dysplasias have been cataloged. In the early seventies there existed no definition and no classification. Freire-Maia and Pinheiro tried to put some order in the field of ectodermal dysplasias. Firstly, the group should be defined before an attempt was made to list its conditions. Secondly, the group was so large that it was necessary to split it into several subgroups. So they decided that an ED should present any two of the signs that affected the four structures widely mentioned by the authors who studied the classic EDs – hair, teeth, nails and sweat glands – with or without any other sign (see blow). The system is arbitrary without biological relevance to the pathogenesis and genetics of the specific disorder. However, classification based on clinical signs and symptoms is all that has been available until recently, since the pathogenesis and molecular genetics of the disorder are largely unknown. Clinical classification of Ectodermal dysplasias (Pinheiro and Freire-Maia, 1994) Unknown cause Conditions AD AR XL ? AD? AR? XL? Subgroup 1-2-3-4 1. Christ-Siemens-Touraine (CST) syndrome (MIM 305100; XR BDE 0333; POS 3208; FMP 1) 2. -

Table I. Genodermatoses with Known Gene Defects 92 Pulkkinen
92 Pulkkinen, Ringpfeil, and Uitto JAM ACAD DERMATOL JULY 2002 Table I. Genodermatoses with known gene defects Reference Disease Mutated gene* Affected protein/function No.† Epidermal fragility disorders DEB COL7A1 Type VII collagen 6 Junctional EB LAMA3, LAMB3, ␣3, 3, and ␥2 chains of laminin 5, 6 LAMC2, COL17A1 type XVII collagen EB with pyloric atresia ITGA6, ITGB4 ␣64 Integrin 6 EB with muscular dystrophy PLEC1 Plectin 6 EB simplex KRT5, KRT14 Keratins 5 and 14 46 Ectodermal dysplasia with skin fragility PKP1 Plakophilin 1 47 Hailey-Hailey disease ATP2C1 ATP-dependent calcium transporter 13 Keratinization disorders Epidermolytic hyperkeratosis KRT1, KRT10 Keratins 1 and 10 46 Ichthyosis hystrix KRT1 Keratin 1 48 Epidermolytic PPK KRT9 Keratin 9 46 Nonepidermolytic PPK KRT1, KRT16 Keratins 1 and 16 46 Ichthyosis bullosa of Siemens KRT2e Keratin 2e 46 Pachyonychia congenita, types 1 and 2 KRT6a, KRT6b, KRT16, Keratins 6a, 6b, 16, and 17 46 KRT17 White sponge naevus KRT4, KRT13 Keratins 4 and 13 46 X-linked recessive ichthyosis STS Steroid sulfatase 49 Lamellar ichthyosis TGM1 Transglutaminase 1 50 Mutilating keratoderma with ichthyosis LOR Loricrin 10 Vohwinkel’s syndrome GJB2 Connexin 26 12 PPK with deafness GJB2 Connexin 26 12 Erythrokeratodermia variabilis GJB3, GJB4 Connexins 31 and 30.3 12 Darier disease ATP2A2 ATP-dependent calcium 14 transporter Striate PPK DSP, DSG1 Desmoplakin, desmoglein 1 51, 52 Conradi-Hu¨nermann-Happle syndrome EBP Delta 8-delta 7 sterol isomerase 53 (emopamil binding protein) Mal de Meleda ARS SLURP-1 -

Prenatal Diagnosis of Skin Diseases
Arch Dis Child: first published as 10.1136/adc.63.10_Spec_No.1175 on 1 October 1988. Downloaded from Archives of Disease in Childhood, 1988, 63, 1175-1178 Current topic Prenatal diagnosis of skin diseases P W SOOTHILL Nuffield Departmenit of Obstetrics and Gynaecology, University of Oxford The possibility of prenatal diagnosis and abortion of invasive prenatal diagnostic procedures. Although abnormal fetuses reassures many couples consider- successful fetal sampling has been described using ing a pregnancy. Some children are conceived only endoscopic or 'blind' techniques, transabdominal because the pregnancy can be terminated if a ultrasound guided needling has replaced these serious uncorrectable abnormality carried in the methods.>" By holding the ultrasound transducer family is passed on. This paper reviews how skin parallel to the proposed path of the needle, it is pos- diseases can be diagnosed prenatally. Space does sible to identify the target, observe the needle enter not permit reference to most of the original papers the skin, and then guide the needle to the target but there are full recent reviews on this subject. -3 through a safe route." This technique is used to obtain amniotic fluid (amniocentesis), chorionic villi Protected by copyright. Techniques of prenatal diagnosis (placental biopsy), fetal blood (cordocentesis), and biopsies of fetal tissues. For fetal skin biopsy, cup- (1) STUDIES OF MATERNAL BLOOD ped biopsy forceps (figure) are passed down the Maternal serum a fetoprotein concentration is used needle, opened, pressed on to the fetal skin, closed, to screen for fetal defects which involve a break in and the forceps are then withdrawn through the fetal surface, such as spina bifida or exomphalos. -

Abstract Case Report
Volume 19 Number 8 August 2013 Case presentation Eccrine hidrocystomas presenting as multiple papules on the cheeks Ahmad S. Jabbar MD1, Keyan Matinpour MD1, Ida Orengo MD1, Paul M. Rodriguez-Waitkus MD2, Abdul H. Diwan MD2, Kiran Motaparthi MD1 Dermatology Online Journal 19 (8): 11 Baylor College of Medicine, Departments of Dermatology1 and Pathology2, Houston, Texas Correspondence: Kiran Motaparthi MD Assistant Professor, Dept. of Dermatology Baylor College of Medicine 1977 Butler Blvd, Ste. E6.200 Houston, TX 77030 Email: [email protected] ABSTRACT Hidrocystomas are common, benign adnexal neoplasms most frequently found on the eyelids, canthi, or periocular areas. Herein, we report a case of multiple hidrocystomas distributed over less common facial areas: cheeks and cutaneous lips. CASE REPORT A 53-year-old woman presented with 10 to 15-year history of multiple asymptomatic papules on the face. Physical exam disclosed approximately 20-30 translucent blue-grey papules on the cheeks, melolabial folds, and the upper cutaneous lip (Figure 1). Shave biopsy was performed. Histopathologic examination revealed unilocular cystic spaces lined by eccrine epithelium with two layers of cuboidal cells (Figures 2 and 3). Figure1. Multiple blue-grey, translucent papules on the cheeks and upper cutaneous lip Figure 2. Eccrine hidrocystoma, low-power view: unilocular cystic cavity Figure 3. Eccrine hidrocystoma, high-power view: cystic cavity lined by two layers of cuboidal-columnar cells DISCUSSION Hidrocystomas (also known as cystadenomas, sudoriferous cysts, and Moll’s gland cysts) are adenomas of the sweat (apocrine or eccrine) glands. They are more common in adult women and can become more prominent in warm environments [4]. -

Oral Manifestation of Genodermatoses L
Journal of Medicine, Radiology, Pathology & Surgery (2017), 4, 22–27 NARRATIVE REVIEW Oral manifestation of genodermatoses L. Kavya, S. Vandana, Swetha Paulose, Vishwanath Rangdhol, W. John Baliah, T. Dhanraj Department of Oral Medicine and Radiology, Indira Gandhi Institute of Dental Sciences, Pudhucherry, India Keywords Abstract Genodermatoses, mutation, oral Genodermatoses are inherited dermatological disorders associated with the structure manifestation and functions of skin and its appendages. Several genodermatoses presenting with Correspondence multisystem involvement lead to increased morbidity and mortality. Dermatological Dr. L. Kavya, Department of Oral Medicine and diseases, besides including the skin and its supplements may also involve the oral cavity, Radiology, Indira Gandhi Institute of Dental which deserves special attention considering that they may be the only presenting sign Sciences, Pudhucherry, India. of these disorders. The important aspect to be noted about these disorders is the rarity Phone: +91-9442902745. of the conditions and lack of awareness among the population which are the major E-mail: [email protected] drawbacks in the early diagnosis and prompt management of these diseases. This review intends to outline various genodermatoses with their characteristic oral manifestations. Received: 11 April 2017; Accepted: 15 May 2017 doi: 10.15713/ins.jmrps.97 Introduction hence, it is simplified under three classifications based on its distinct features. Genodermatoses or genetic diseases are a group of inherited According to William et al. 2005:[3] skin disorders with a collection of cutaneous and systemic signs 1. Chromosomal and symptoms. In the oral cavity, a wide spectrum of diseases 2. Single gene occurs due to genetic modifications ranging from developmental 3. -
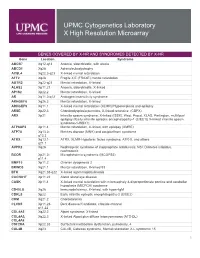
Genes Covered and Disorders Detected by X-HR Microarray
UPMC Cytogenetics Laboratory X High Resolution Microarray GENES COVERED BY X-HR AND SYNDROMES DETECTED BY X-HR Gene Location Syndrome ABCB7 Xq12-q13 Anemia, sideroblastic, with ataxia ABCD1 Xq28 Adrenoleukodystrophy ACSL4 Xq22.3-q23 X-linked mental retardation AFF2 Xq28 Fragile X E (FRAXE) mental retardation AGTR2 Xq22-q23 Mental retardation, X-linked ALAS2 Xp11.21 Anemia, sideroblastic, X-linked AP1S2 Xp22.2 Mental retardation, X-linked AR Xq11.2-q12 Androgen insensitivity syndrome ARHGEF6 Xq26.3 Mental retardation, X-linked ARHGEF9 Xq11.1 X-linked mental retardation (XLMR)//Hyperekplexia and epilepsy ARSE Xp22.3 Chondrodysplasia punctata, X-linked recessive (CDPX) ARX Xp21 Infantile spasm syndrome, X-linked (ISSX), West, Proud, XLAG, Partington, multifocal epilepsy //Early infantile epileptic encephalopathy-1 (EIEE1)( X-linked infantile spasm syndrome-1-ISSX1) ATP6AP2 Xp11.4 Mental retardation, X-linked, with epilepsy (XMRE) ATP7A Xq13.2- Menkes disease (MNK) and occipital horn syndrome q13.3 ATRX Xq13.1- ATRX, XLMR-Hypotonic facies syndrome, ATR-X, and others q21.1 AVPR2 Xq28 Nephrogenic syndrome of inappropriate antidiuresis; NSI; Diabetes insipidus, nephrogenic BCOR Xp21.2- Microphthalmia syndromic (MCOPS2) p11.4 BMP15 Xp11.2 Ovarian dysgenesis 2 BRWD3 Xq21.1 Mental retardation, X-linked 93 BTK Xq21.33-q22 X-linked agammaglobulinemia CACNA1F Xp11.23 Aland Island eye disease CASK Xp11.4 X-linked mental retardation with microcephaly & disproportionate pontine and cerebellar hypoplasia (MICPCH) syndrome CD40LG Xq26 Immunodeficiency, -

NGS Oncology)
UNIVERSITY OF MINNESOTA PHYSICIANS OUTREACH LABS Submit this form along with the appropriate Molecular requisition (Molecular Diagnostics or MOLECULAR DIAGNOSTICS (612) 273-8445 DATE: TIME COLLECTED: PCU/CLINIC: Molecular NGS Oncology). AM PM PATIENT IDENTIFICATION DIAGNOSIS (Dx) / DIAGNOSIS CODES (ICD-9) - OUTPATIENTS ONLY SPECIMEN TYPE: o Blood (1) (2) (3) (4) PLEASE COLLECT 5-10CC IN ACD-A OR EDTA TUBE ORDERING PHYSICIAN NAME AND PHONE NUMBER: Tests can be ordered as a full panel, or by individual gene(s). Please contact the genetic counselor with any questions at 612-624-8948 or by pager at 612-899-3291. _______________________________________________ Test Ordered- EPIC: Next generation sequencing(Next Gen) Sunquest: NGS Ectodermal dysplasia epidermolysis bullosa simplex with Acne inversa muscular dystrophy Full panel PLEC Full panel EDA Epidermolytic hyperkeratosis NCSTN EDARADD Full panel PSENEN MSX1 KRT1 PSEN1 KRT85 KRT10 Acrodermatitis enteropathica PVRL4 Erythroderma, congenital, with NFKBIA palmoplantar keratoderma, SLC39A4 IKBKG hypotrichosis, and hyper IgE Amyloidosis, primary localized Ectodermal dysplasia/skin fragility DSG1 cutaneous, 1 syndrome Erythrokeratodermia variabilis with PKP1 erythema gyratum repens Full panel Ectrodactyly, ectodermal dysplasia, GJB4 OSMR and cleft lip/palate syndrome 3 Familial benign pemphigus IL31RA TP63 ATP2C1 Atrichia with papular lesions Focal facial dermal dysplasia 3 Focal dermal hypoplasia TWIST2 HR PORCN Epidermodysplasia verruciformis Autosomal recessive hypohidrotic Glomuvenous malformations -

17Th ESPD Annual Meeting 19 – 21 October 2017 Meliá Palas Atenea Palma De Mallorca, Spain
ESPD 2017 PALMA DE MALLORCA 17th ESPD Annual Meeting 19 – 21 October 2017 Meliá Palas Atenea Palma de Mallorca, Spain List of Posters www.espd.info Posters DIAGNOSIS P 001 | COLOR DOPPLER ULTRASOUND: EXPERIENCE OF ITS USEFULNESS IN PEDIATRIC PATIENTS Giavedoni, P.; Morgado-Carrasco, D.; Carrera, C.; Ferrando, J. (Spain) P 002 | ULTRASOUND FINDINGS IN IDIOPATHIC ASEPTIC FACIAL GRANULOMA Gómez-Zubiaur, A.; Knöpfel, N.; Noguera-Morel, L.; Cabrera-Hernández, A.; Torrelo, A.; Hernández-Martín, Á. (Spain) P 003 | HALO SCALP RING. ULTRASONOGRAPHIC FINDINGS Macías del Toro, E.; Torre Castro, J.; Nuñez Hipolito, L.; Lopez Robles, J.; Mendoza Cembranos, M.D.; Alfageme Roldan, F.; Cabeza Martínez, R.; Gonzalez de Domingo, M.A.; Dolores, S.M.; Roustan Gullon, G. (Spain) P 004 | EPIDEMIOLOGY AND OUTCOME ANALYSIS OF 395 INFANTS GETTING DERMATOLOGIC SURGERY ATTENDING A DEPARTMENT OF DERMATOLOGY IN KOREA Park, K.; Kim, J.H.; Im, B.R. (Republic of Korea) P 005 | SONOGRAPHIC FINDINGS IN SUBCUTANEOUS GRANULOMA ANNULARE Rodríguez Díaz, E.; Quevedo, A.; González Díaz, M.E.; Rodríguez Vidal, A.; González-Sánchez, S.; García Suárez, L.; Vázquez Osorio, I. (Spain) GENODERMATOSIS P 006 | A CASE OF EPIDERMOLYTIC ICHTHYOSIS WITH A DE NOVO KRT1 GENE MUTATION Abdelrahman, W.; Clements, S.; Hoey, S. (United Kingdom) P 007 | AN UNUSUAL CAUSE OF FACIAL ULCERATION IN A FOUR YEAR OLD Abdelrahman, W.; Armstrong, K. (United Kingdom) P 008 | GOLTZ SYNDROME: REPORT OF TWO CASES AND OVERVIEW OF PORCN MUTATIONS Akkaya, A.D.; Özlü, C.; Zeynep, E.; Umut, A.; Azaklı, H.; Eraslan, S.; Kayserili, H. (Turkey) P 009 | GOLTZ SYNDROME: CASE REPORT OF FOCAL DERMAL HYPOPLASIA Alrehaili, D.; Alshihry, H.; Alharthi, N.; (Saudi Arabia) P 010 | TWO CASES OF INCONTINENTIA PIGMENTI IN NEWBORNS WITH CONSEQUENT VISUAL ABNORMALITIES Alwash, N.; Dinani, N.; Felton, J. -

1 – Ust-Dzhegutinsky District; 2
Supplementary Tables S1–S6 Note: 1 – Ust-Dzhegutinsky district; 2 – Karachaevsky district; 3 – Malokarachaevsky district; 4– Cherkessk City; 5 – Prikubansky district; 6 – Urupsky district; 7 – Zelenchuksky district; 8 – Abazinsky district; 9 – Khabezsky district; 10 – Adyge-Khablsky district; 11 – Nogaysky district; T/I – type of inheritance; AD – autosomal dominant type of inheritance, AR – autosomal recessive type of inheritance, XL – X-linked type of inheritance. PS – Phenotypic Series for OMIM in case of heterogeneity of the disease; Isol. – Isolated cases; Som. – Somatic mutation Table S1. Nosological spectrum and prevalence (per 100000) of hereditary neurological diseases in Karachay-Cherkess Republic (KChR) Number of patients Prevalence (per 100000) 1 2 3 4 5 6 7 8 9 10 11 Σ Kara Russi Circ Aba Nog Other Σ № ОМIМ Diagnosis T/I chays an assia zins ais s ns 1. #156200 Undifferentiated mental retardation АD 7 4 4 7 4 6 7 39 12.31 6.68 15.7 13.6 9.50 2. #249500 Undifferentiated mental retardation AR 10 10 19 6 23 9 1 2 17 16 20 133 31.40 26.71 41.2 9.02 74.6 76.68 32.41 3. #309530 Undifferentiated mental retardation XL 10 8 3 6 18 5 6 3 12 2 3 76 40.63 26.71 19.7 42.1 54.3 125.5 37.04 4. #300624 Martin-Bell syndrome XL 2 1 4 3 2 2 1 15 8.62 7.87 36.1 7.31 5. #158600 Spinal muscular atrophy, juvenile, proximal АD 2 2 1.48 0.49 6. #253300 Spinal muscular atrophy, type 1 AR 1 1 2 0.62 3.01 0.49 7.