Genetic and Functional Evidence for a Locus Controlling Otitis Media At
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Meta-Analysis of Nasopharyngeal Carcinoma
BMC Genomics BioMed Central Research article Open Access Meta-analysis of nasopharyngeal carcinoma microarray data explores mechanism of EBV-regulated neoplastic transformation Xia Chen†1,2, Shuang Liang†1, WenLing Zheng1,3, ZhiJun Liao1, Tao Shang1 and WenLi Ma*1 Address: 1Institute of Genetic Engineering, Southern Medical University, Guangzhou, PR China, 2Xiangya Pingkuang associated hospital, Pingxiang, Jiangxi, PR China and 3Southern Genomics Research Center, Guangzhou, Guangdong, PR China Email: Xia Chen - [email protected]; Shuang Liang - [email protected]; WenLing Zheng - [email protected]; ZhiJun Liao - [email protected]; Tao Shang - [email protected]; WenLi Ma* - [email protected] * Corresponding author †Equal contributors Published: 7 July 2008 Received: 16 February 2008 Accepted: 7 July 2008 BMC Genomics 2008, 9:322 doi:10.1186/1471-2164-9-322 This article is available from: http://www.biomedcentral.com/1471-2164/9/322 © 2008 Chen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Epstein-Barr virus (EBV) presumably plays an important role in the pathogenesis of nasopharyngeal carcinoma (NPC), but the molecular mechanism of EBV-dependent neoplastic transformation is not well understood. The combination of bioinformatics with evidences from biological experiments paved a new way to gain more insights into the molecular mechanism of cancer. Results: We profiled gene expression using a meta-analysis approach. Two sets of meta-genes were obtained. Meta-A genes were identified by finding those commonly activated/deactivated upon EBV infection/reactivation. -

Supplemental Figure and Table Legends
Supplemental figure and table legends Supplementary Figure 1: KIAA1841 is well conserved among vertebrates. NCBI HomoloGene pairwise alignment scores of human KIAA1841 sequence compared to other vertebrate orthologs. Supplementary Figure 2: µ-germline transcripts (GLT) and AID mRNA expression are not affected by overexpression of KIAA1841. Splenic B cells were isolated from wild-type mice, and transduced with retroviral vector control (pMIG) or a vector expressing KIAA1841. Levels of µ-GLT and AID mRNA were determined at 72h post-infection by RT-qPCR, and normalized to -actin mRNA and the pMIG control. The mean of three independent experiments +/- SD is shown. NS, p = not significant, p 0.05, two-tailed paired student’s t-test. Supplementary Figure 3: Overexpression of untagged and Xpress-tagged KIAA1841 does not affect cell proliferation. Splenic B cells were isolated from wild-type mice, stimulated with LPS+IL4, and transduced with retroviral vector control (pMIG) or a vector expressing KIAA1841 or Xpress (Xp)-tagged KIAA1841. Cells are labeled with seminaphthorhodafluor (SNARF) cell tracking dye and SNARF intensity was measured at 0h, 24h, and 48h after retroviral infection. Histograms of transduced cells (GFP+) for pMIG control, KIAA1841 and Xp-KIAA1841 were superimposed at each time point. Three independent retroviral infection experiments are shown. Supplementary Figure 4: Sequence alignment of the putative SANT domain of KIAA1841 with the SANT domain of SWI3. Alignment was performed using ClustalOmega; *, conserved residue, :, strongly similar residues, ., weakly similar residues. Numbers indicate amino acid residues in each sequence. Helix 3, which has been reported to be important for the chromatin remodeling function of SWI3 (Boyer et. -

Supplementary Material Contents
Supplementary Material Contents Immune modulating proteins identified from exosomal samples.....................................................................2 Figure S1: Overlap between exosomal and soluble proteomes.................................................................................... 4 Bacterial strains:..............................................................................................................................................4 Figure S2: Variability between subjects of effects of exosomes on BL21-lux growth.................................................... 5 Figure S3: Early effects of exosomes on growth of BL21 E. coli .................................................................................... 5 Figure S4: Exosomal Lysis............................................................................................................................................ 6 Figure S5: Effect of pH on exosomal action.................................................................................................................. 7 Figure S6: Effect of exosomes on growth of UPEC (pH = 6.5) suspended in exosome-depleted urine supernatant ....... 8 Effective exosomal concentration....................................................................................................................8 Figure S7: Sample constitution for luminometry experiments..................................................................................... 8 Figure S8: Determining effective concentration ......................................................................................................... -

Comprehensive Transcriptome Analysis of Mouse Embryonic Stem Cell Adipogenesis Unravels New Processes of Adipocyte Development
Billon et al. Genome Biology 2010, 11:R80 http://genomebiology.com/2010/11/8/R80 RESEARCH Open Access Comprehensive transcriptome analysis of mouse embryonic stem cell adipogenesis unravels new processes of adipocyte development Nathalie Billon1*, Raivo Kolde2,3†, Jüri Reimand2†, Miguel C Monteiro1†, Meelis Kull2,3, Hedi Peterson3,4, Konstantin Tretyakov2, Priit Adler4, Brigitte Wdziekonski1, Jaak Vilo2,3, Christian Dani1 Abstract Background: The current epidemic of obesity has caused a surge of interest in the study of adipose tissue formation. While major progress has been made in defining the molecular networks that control adipocyte terminal differentiation, the early steps of adipocyte development and the embryonic origin of this lineage remain largely unknown. Results: Here we performed genome-wide analysis of gene expression during adipogenesis of mouse embryonic stem cells (ESCs). We then pursued comprehensive bioinformatic analyses, including de novo functional annotation and curation of the generated data within the context of biological pathways, to uncover novel biological functions associated with the early steps of adipocyte development. By combining in-depth gene regulation studies and in silico analysis of transcription factor binding site enrichment, we also provide insights into the transcriptional networks that might govern these early steps. Conclusions: This study supports several biological findings: firstly, adipocyte development in mouse ESCs is coupled to blood vessel morphogenesis and neural development, just as it is during mouse development. Secondly, the early steps of adipocyte formation involve major changes in signaling and transcriptional networks. A large proportion of the transcription factors that we uncovered in mouse ESCs are also expressed in the mouse embryonic mesenchyme and in adipose tissues, demonstrating the power of our approach to probe for genes associated with early developmental processes on a genome-wide scale. -

Universidad Autónoma De Madrid Programa De
UNIVERSIDAD AUTÓNOMA DE MADRID PROGRAMA DE DOCTORADO EN BIOCIENCIAS MOLECULARES FUNCTIONAL RELEVANCE OF PP2A-B55 PHOSPHATASES IN THE MAMMALIAN CELL CYCLE TESIS DOCTORAL María Sanz Flores Madrid, 2017 DEPARTAMENTO DE BIOLOGÍA MOLECULAR FACULTAD DE CIENCIAS UNIVERSIDAD AUTÓNOMA DE MADRID FUNCTIONAL RELEVANCE OF PP2A-B55 PHOSPHATASES IN THE MAMMALIAN CELL CYCLE María Sanz Flores Licenciada en Biología Director: Marcos Malumbres Martínez Codirectora: Mónica Álvarez Fernández Centro Nacional de Investigaciones Oncológicas (CNIO) Madrid, 2017 Mónica Álvarez Fernández, Investigadora del Grupo de División Celular y Cáncer del Centro Nacional de Investigaciones Oncológicas (CNIO) y, Marcos Malumbres Martínez, Jefe del Grupo de División Celular y Cáncer del Centro Nacional de Investigaciones Oncológicas (CNIO) Certifican: que María Sanz Flores ha realizado bajo su dirección el trabajo de Tesis Doctoral titulado: FUNCTIONAL RELEVANCE OF PP2A-B55 PHOSPHATASES IN THE MAMMALIAN CELL CYCLE Revisado el presente trabajo, consideran que reúne todos los méritos necesarios para su presentación y defensa con el fin de optar al grado de Doctor por la Universidad Autónoma de Madrid. Mónica Álvarez Fernández Marcos Malumbres RESUMEN/SUMMARY Resumen RESUMEN La fosforilación reversible de proteínas es un mecanismo esencial en la regulación del ciclo celular. Mientras que el papel llevado a cabo en mitosis por las proteínas quinasa ha sido profundamente caracterizado, la identidad y la función específica de las fosfatasas en la mitosis de mamíferos está aún por determinar. La proteína fosfatasa 2A (PP2A) es una fosfatasa de residuos serina y treonina de gran importancia en células eucarióticas. Existen evidencias de que los complejos formados por PP2A con subunidades reguladoras de la familia B55 juegan un importante papel en la desfosforilación de sustratos de quinasas dependiente de ciclina (CDKs) durante mitosis en diferentes organismos. -

NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 11-7-2018 Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer Jairo Ramos [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Clinical Epidemiology Commons Recommended Citation Ramos, Jairo, "Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer" (2018). FIU Electronic Theses and Dissertations. 3872. https://digitalcommons.fiu.edu/etd/3872 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida DECIPHER MECHANISMS BY WHICH NUCLEAR RESPIRATORY FACTOR ONE (NRF1) COORDINATES CHANGES IN THE TRANSCRIPTIONAL AND CHROMATIN LANDSCAPE AFFECTING DEVELOPMENT AND PROGRESSION OF INVASIVE BREAST CANCER A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in PUBLIC HEALTH by Jairo Ramos 2018 To: Dean Tomás R. Guilarte Robert Stempel College of Public Health and Social Work This dissertation, Written by Jairo Ramos, and entitled Decipher Mechanisms by Which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer, having been approved in respect to style and intellectual content, is referred to you for judgment. -
Primepcr™Assay Validation Report
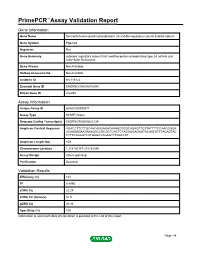
PrimePCR™Assay Validation Report Gene Information Gene Name Serine/threonine-protein phosphatase 2A 55 kDa regulatory subunit B delta isoform Gene Symbol Ppp2r2d Organism Rat Gene Summary cytosolic regulatory subunit that modifies protein phosphatase type 2A activity and subcellular localization Gene Aliases Not Available RefSeq Accession No. Not Available UniGene ID Rn.116122 Ensembl Gene ID ENSRNOG00000016940 Entrez Gene ID 246255 Assay Information Unique Assay ID qRnoCID0008377 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000023139 Amplicon Context Sequence AGACCTTCTTGCAACAGGAGACAAAGGTGGCAGAGTTGTTATTTTCCAACGGGA ACAAGAGAATAAAGGCCGCGCTCACTCTAGGGGAGAGTACAATGTTTACAGTAC CTTTCAGAGTCATGAGCCAGAGTTTGACTAT Amplicon Length (bp) 109 Chromosome Location 1:218152187-218153346 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 101 R2 0.9995 cDNA Cq 22.29 cDNA Tm (Celsius) 81.5 gDNA Cq 36.36 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Ppp2r2d, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

An Expanded Proteome of Cardiac T-Tubules☆
Cardiovascular Pathology 42 (2019) 15–20 Contents lists available at ScienceDirect Cardiovascular Pathology Original Article An expanded proteome of cardiac t-tubules☆ Jenice X. Cheah, Tim O. Nieuwenhuis, Marc K. Halushka ⁎ Department of Pathology, Division of Cardiovascular Pathology, Johns Hopkins University SOM, Baltimore, MD, USA article info abstract Article history: Background: Transverse tubules (t-tubules) are important structural elements, derived from sarcolemma, found Received 27 February 2019 on all striated myocytes. These specialized organelles create a scaffold for many proteins crucial to the effective Received in revised form 29 April 2019 propagation of signal in cardiac excitation–contraction coupling. The full protein composition of this region is un- Accepted 17 May 2019 known. Methods: We characterized the t-tubule subproteome using 52,033 immunohistochemical images covering Keywords: 13,203 proteins from the Human Protein Atlas (HPA) cardiac tissue microarrays. We used HPASubC, a suite of Py- T-tubule fi Proteomics thon tools, to rapidly review and classify each image for a speci c t-tubule staining pattern. The tools Gene Cards, Caveolin String 11, and Gene Ontology Consortium as well as literature searches were used to understand pathways and relationships between the proteins. Results: There were 96 likely t-tubule proteins identified by HPASubC. Of these, 12 were matrisome proteins and 3 were mitochondrial proteins. A separate literature search identified 50 known t-tubule proteins. A comparison of the 2 lists revealed only 17 proteins in common, including 8 of the matrisome proteins. String11 revealed that 94 of 127 combined t-tubule proteins generated a single interconnected network. Conclusion: Using HPASubC and the HPA, we identified 78 novel, putative t-tubule proteins and validated 17 within the literature. -

Live-Cell Imaging Rnai Screen Identifies PP2A–B55α and Importin-Β1 As Key Mitotic Exit Regulators in Human Cells
LETTERS Live-cell imaging RNAi screen identifies PP2A–B55α and importin-β1 as key mitotic exit regulators in human cells Michael H. A. Schmitz1,2,3, Michael Held1,2, Veerle Janssens4, James R. A. Hutchins5, Otto Hudecz6, Elitsa Ivanova4, Jozef Goris4, Laura Trinkle-Mulcahy7, Angus I. Lamond8, Ina Poser9, Anthony A. Hyman9, Karl Mechtler5,6, Jan-Michael Peters5 and Daniel W. Gerlich1,2,10 When vertebrate cells exit mitosis various cellular structures can contribute to Cdk1 substrate dephosphorylation during vertebrate are re-organized to build functional interphase cells1. This mitotic exit, whereas Ca2+-triggered mitotic exit in cytostatic-factor- depends on Cdk1 (cyclin dependent kinase 1) inactivation arrested egg extracts depends on calcineurin12,13. Early genetic studies in and subsequent dephosphorylation of its substrates2–4. Drosophila melanogaster 14,15 and Aspergillus nidulans16 reported defects Members of the protein phosphatase 1 and 2A (PP1 and in late mitosis of PP1 and PP2A mutants. However, the assays used in PP2A) families can dephosphorylate Cdk1 substrates in these studies were not specific for mitotic exit because they scored pro- biochemical extracts during mitotic exit5,6, but how this relates metaphase arrest or anaphase chromosome bridges, which can result to postmitotic reassembly of interphase structures in intact from defects in early mitosis. cells is not known. Here, we use a live-cell imaging assay and Intracellular targeting of Ser/Thr phosphatase complexes to specific RNAi knockdown to screen a genome-wide library of protein substrates is mediated by a diverse range of regulatory and targeting phosphatases for mitotic exit functions in human cells. We subunits that associate with a small group of catalytic subunits3,4,17. -

Anti-MASTL Monoclonal Antibody, Clone SJQMZ 85D (DCABH-4990) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-MASTL monoclonal antibody, clone SJQMZ 85D (DCABH-4990) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Product Overview Mouse monoclonal to MASTL Antigen Description Serine/threonine kinase that plays a key role in M phase by acting as a regulator of mitosis entry and maintenance. Acts by promoting the inactivation of protein phosphatase 2A (PP2A) during M phase: does not directly inhibit PP2A but acts by mediating phosphorylation and subsequent activation of ARPP19 and ENSA at Ser-62 and Ser-67, respectively. ARPP19 and ENSA are phosphatase inhibitors that specifically inhibit the PPP2R2D (PR55-delta) subunit of PP2A. Inactivation of PP2A during M phase is essential to keep cyclin-B1-CDK1 activity high. Following DNA damage, it is also involved in checkpoint recovery by being inhibited. Phosphorylates histone protein in vitro; however such activity is unsure in vivo. May be involved in megakaryocyte differentiation. Immunogen Recombinant full length protein, corresponding to amino acids 1-879 of Human MASTL with a 26kDa proprietary tag. Isotype IgG1 Source/Host Mouse Species Reactivity Mouse, Human Clone SJQMZ 85D Purity Protein G purified Conjugate Unconjugated Applications WB, IHC-P Positive Control Human and Mouse MASTL-transfected HEK cell lysates; Raji, Ramos, Dana and MT2 cell lysates; 293T and HeLa transfected cell lysates; Human tonsil, testicle, bone marrow, Hodgkin and Burkitt lymphoma tissues. Format Liquid Size 100 μg Buffer pH: 7.40; Constituents: PBS, 1% BSA 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Preservative None Storage Store at +4°C short term (1-2 weeks). -

UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Cardiac Stretch-Induced Transcriptomic Changes are Axis-Dependent Permalink https://escholarship.org/uc/item/7m04f0b0 Author Buchholz, Kyle Stephen Publication Date 2016 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Cardiac Stretch-Induced Transcriptomic Changes are Axis-Dependent A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioengineering by Kyle Stephen Buchholz Committee in Charge: Professor Jeffrey Omens, Chair Professor Andrew McCulloch, Co-Chair Professor Ju Chen Professor Karen Christman Professor Robert Ross Professor Alexander Zambon 2016 Copyright Kyle Stephen Buchholz, 2016 All rights reserved Signature Page The Dissertation of Kyle Stephen Buchholz is approved and it is acceptable in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California, San Diego 2016 iii Dedication To my beautiful wife, Rhia. iv Table of Contents Signature Page ................................................................................................................... iii Dedication .......................................................................................................................... iv Table of Contents ................................................................................................................ v List of Figures ...................................................................................................................