Riley Oxidation of Heterocyclic Intermediates on Paths to Hydroporphyrins—A Review
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
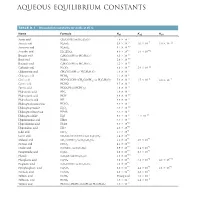
Aqueous Equilibrium Constants
BLB_APP_D_1062-1063hr.qxp 11/8/10 2:27 PM Page 1062 APPENDIX D AQUEOUS EQUILIBRIUM CONSTANTS TABLE D.1 • Dissociation Constants for Acids at 25 ˚C Name Formula Ka1 Ka2 Ka3 -5 Acetic acid CH3COOH (or HC2H3O2) 1.8 * 10 * -3 * -7 * -12 Arsenic acid H3AsO4 5.6 10 1.0 10 3.0 10 * -10 Arsenous acid H3AsO3 5.1 10 * -5 * -12 Ascorbic acid H2C6H6O6 8.0 10 1.6 10 * -5 Benzoic acid C6H5COOH (or HC7H5O2) 6.3 10 * -10 Boric acid H3BO3 5.8 10 * -5 Butanoic acid C3H7COOH (or HC4H7O2) 1.5 10 * -7 * -11 Carbonic acid H2CO3 4.3 10 5.6 10 * -3 Chloroacetic acid CH2ClCOOH (or HC2H2O2Cl) 1.4 10 * -2 Chlorous acid HClO2 1.1 10 * -4 * -5 -7 Citric acid HOOCC(OH) (CH2COOH)2 (or H3C6H5O7) 7.4 10 1.7 10 4.0 * 10 - Cyanic acid HCNO 3.5 * 10 4 * -4 Formic acid HCOOH (or HCHO2) 1.8 10 * -5 Hydroazoic acid HN3 1.9 10 - Hydrocyanic acid HCN 4.9 * 10 10 - Hydrofluoric acid HF 6.8 * 10 4 - -7 Hydrogen chromate ion HCrO4 3.0 * 10 * -12 Hydrogen peroxide H2O2 2.4 10 - -2 Hydrogen selenate ion HSeO4 2.2 * 10 * -8 * -19 Hydrogen sulfide H2S 9.5 10 1 10 - Hypobromous acid HBrO 2.5 * 10 9 - Hypochlorous acid HClO 3.0 * 10 8 - Hypoiodous acid HIO 2.3 * 10 11 * -1 Iodic acid HIO3 1.7 10 * -4 Lactic acid CH3CH(OH)COOH (or HC3H5O3) 1.4 10 * -3 * -6 Malonic acid CH2(COOH)2 (or H2C3H2O4) 1.5 10 2.0 10 * -4 Nitrous acid HNO2 4.5 10 * -2 * -5 Oxalic acid (COOH)2 (or H2C2O4) 5.9 10 6.4 10 * -2 * -9 Paraperiodic acid H5IO6 2.8 10 5.3 10 * -10 Phenol C6H5OH (or HC6H5O) 1.3 10 * -3 * -8 * -13 Phosphoric acid H3PO4 7.5 10 6.2 10 4.2 10 * -5 Propionic acid C2H5COOH (or HC3H5O2) 1.3 10 * -

Zn-Nx Sites on N-Doped Carbon for Aerobic Oxidative Cleavage
ARTICLE https://doi.org/10.1038/s41467-021-25118-0 OPEN Zn-Nx sites on N-doped carbon for aerobic oxidative cleavage and esterification of C(CO)-C bonds ✉ ✉ Chao Xie1, Longfei Lin 2, Liang Huang 3, Zixin Wang1, Zhiwei Jiang 1, Zehui Zhang1 & Buxing Han 2 Selective cleavage of C-C bonds is very important in organic chemistry, but remains chal- lenging because of their inert chemical nature. Herein, we report that Zn/NC-X catalysts, in 1234567890():,; which Zn2+ coordinate with N species on microporous N-doped carbon (NC) and X denotes the pyrolysis temperature, can effectively catalyze aerobic oxidative cleavage of C(CO)-C bonds and quantitatively convert acetophenone to methyl benzoate with a yield of 99% at 100 °C. The Zn/NC-950 can be applied for a wide scope of acetophenone derivatives as well as more challenging alkyl ketones. Detail mechanistic investigations reveal that the catalytic performance of Zn/NC-950 can be attributed to the coordination between Zn2+ and N species to change the electronic state of the metal, synergetic effect of the Zn single sites with their surrounding N atoms, as well as the microporous structure with the high surface area and structural defects of the NC. 1 Key Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education & Hubei Key Laboratory of Catalysis and Materials Science, South- Central University for Nationalities, Wuhan, China. 2 Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid, Interface and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing, China. 3 The State Key Laboratory of Refractories and Metallurgy, ✉ Wuhan University of Science and Technology, Wuhan, China. -

Synthesis and Consecutive Reactions of Α-Azido Ketones: a Review
Molecules 2015, 20, 14699-14745; doi:10.3390/molecules200814699 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Synthesis and Consecutive Reactions of α-Azido Ketones: A Review Sadia Faiz 1,†, Ameer Fawad Zahoor 1,*, Nasir Rasool 1,†, Muhammad Yousaf 1,†, Asim Mansha 1,†, Muhammad Zia-Ul-Haq 2,† and Hawa Z. E. Jaafar 3,* 1 Department of Chemistry, Government College University Faisalabad, Faisalabad-38000, Pakistan, E-Mails: [email protected] (S.F.); [email protected] (N.R.); [email protected] (M.Y.); [email protected] (A.M.) 2 Office of Research, Innovation and Commercialization, Lahore College for Women University, Lahore-54600, Pakistan; E-Mail: [email protected] 3 Department of Crop Science, Faculty of Agriculture, Universiti Putra Malaysia, Serdang-43400, Selangor, Malaysia † These authors contributed equally to this work. * Authors to whom correspondence should be addressed; E-Mails: [email protected] (A.F.Z.); [email protected] (H.Z.E.J.); Tel.: +92-333-6729186 (A.F.Z.); Fax: +92-41-9201032 (A.F.Z.). Academic Editors: Richard A. Bunce, Philippe Belmont and Wim Dehaen Received: 20 April 2015 / Accepted: 3 June 2015 / Published: 13 August 2015 Abstract: This review paper covers the major synthetic approaches attempted towards the synthesis of α-azido ketones, as well as the synthetic applications/consecutive reactions of α-azido ketones. Keywords: α-azido ketones; synthetic applications; heterocycles; click reactions; drugs; azides 1. Introduction α-Azido ketones are very versatile and valuable synthetic intermediates, known for their wide variety of applications, such as in amine, imine, oxazole, pyrazole, triazole, pyrimidine, pyrazine, and amide alkaloid formation, etc. -

Acids and Bases
Name Date Class CHAPTER 14 REVIEW Acids and Bases SECTION 1 SHORT ANSWER Answer the following questions in the space provided. 1. Name the following compounds as acids: sulfuric acid a. H2SO4 sulfurous acid b. H2SO3 hydrosulfuric acid c. H2S perchloric acid d. HClO4 hydrocyanic acid e. hydrogen cyanide 2. H2S Which (if any) of the acids mentioned in item 1 are binary acids? 3. Write formulas for the following acids: HNO2 a. nitrous acid HBr b. hydrobromic acid H3PO4 c. phosphoric acid CH3COOH d. acetic acid HClO e. hypochlorous acid 4. Calcium selenate has the formula CaSeO4. H2SeO4 a. What is the formula for selenic acid? H2SeO3 b. What is the formula for selenous acid? 5. Use an activity series to identify two metals that will not generate hydrogen gas when treated with an acid. Choose from Cu, Ag, Au, Pt, Pd, or Hg. 6. Write balanced chemical equations for the following reactions of acids and bases: a. aluminum metal with dilute nitric acid ϩ → ϩ 2Al(s) 6HNO3(aq) 2Al(NO3)3(aq) 3H2(g) b. calcium hydroxide solution with acetic acid ϩ → ϩ Ca(OH)2(aq) 2CH3COOH(aq) Ca(CH3COO)2(aq) 2H2O(l ) MODERN CHEMISTRY ACIDS AND BASES 117 Copyright © by Holt, Rinehart and Winston. All rights reserved. Name Date Class SECTION 1 continued 7. Write net ionic equations that represent the following reactions: a. the ionization of HClO3 in water ϩ → ϩ ϩ Ϫ HClO3(aq) H2O(l ) H3O (aq) ClO3 (aq) b. NH3 functioning as an Arrhenius base ϩ → ϩ ϩ Ϫ NH3(aq) H2O(l ) ← NH4 (aq) OH (aq) 8. -

Carboligation Using the Aldol Reaction
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1730 Carboligation using the aldol reaction A comparison of stereoselectivity and methods DERAR AL-SMADI ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 ISBN 978-91-513-0472-4 UPPSALA urn:nbn:se:uu:diva-362866 2018 Dissertation presented at Uppsala University to be publicly examined in BMC C2:301, Husargatan 3, Uppsala, Friday, 30 November 2018 at 09:15 for the degree of Doctor of Philosophy. The examination will be conducted in English. Faculty examiner: Professor Ulf Nilsson (Lund University). Abstract Al-Smadi, D. 2018. Carboligation using the aldol reaction. A comparison of stereoselectivity and methods. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1730. 50 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-513-0472-4. The research summarized in this thesis focuses on synthesizing aldehyde and aldol compounds as substrates and products for the enzyme D-fructose-6-aldolase (FSA). Aldolases are important enzymes for the formation of carbon-carbon bonds in nature. In biological systems, aldol reactions, both cleavage and formation play central roles in sugar metabolism. Aldolases exhibit high degrees of stereoselectivity and can steer the product configurations to a given enantiomeric and diastereomeric form. To become truly useful synthetic tools, the substrate scope of these enzymes needs to become broadened. In the first project, phenylacetaldehyde derivatives were synthesized for the use as test substrates for E. coli FSA. Different methods were discussed to prepare phenylacetaldehyde derivatives, the addition of a one carbon unit to benzaldehyde derivatives using a homologation reaction was successful and was proven efficient and non-sensitive to the moisture. -

Process for the Preparation of Chacogens and Chalcogenide Alloys of Controlled Average Crystallite Size
Europaisches Patentamt European Patent Office @ Publication number: 0 1 60 493 Office europeen des brevets A2 © EUROPEAN PATENT APPLICATION © Application number: 85302825.6 © Int. CI.4: C 01 B 19/02 _ C 01 B 19/04 © Date of filing: 23.04.85 © Priority: 23.04.84 US 603019 © Applicant: XEROX CORPORATION Xerox Square - 020 Rochester New York 14644(US) @ Date of publication of application: 06.11.85 Bulletin 85/45 @ Inventor: Badesha, Santokh Singh 665 Cievenger Road @ Designated Contracting States: Ontario New York 1451 9(US) DE FR GB © Inventor: Fekete, George Thomas 111 Dale Road Rochester New York 14625IUS) © Representative: Goode, Ian Roy et al, European Patent Attorney do Rank Xerox Limited Patent Department Rank Xerox House 338 Euston Road London NW1 3BH(GB) (64) Process for the preparation of chacogens and chalcogenide alloys of controlled average crystallite size. A process for the preparation of chalcogens and chal- cogenide alloys which comprises subjecting the correspond- ing esters to a reduction or coreduction reaction, for example using hydrazine, at a temperature between about 40°C and 135°C. The morphology/crystallite size of the product is controlled by suitably selecting a temperature within this range, depending on the particular chalcogen or alloy being prepared. The product is either a non-crystalline material, or has an average crystallite size between about 15 and 52 nm. This invention relates to a process for the preparation of chalcogens and chalcogenide alloys, the process being of the kind which comprises subjecting the corresponding esters to a reduction or coreduction reaction. Such a process for preparing high purity tellurium is described in EP-A-0 102 753, and such a process for preparing high purity chalcogenide alloys, such as selenium/tellurium alloys, is described in EP-A-0 101 238. -

1,2-Cyclohexanedione Dioxime
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

H2seo3. Arxiv:1901.03765V1
Crystal Structure Elucidation of the Novel Molecular-Inorganic Polymer K2SeO4 · H2SeO3. Oscar S. Hernández-Daguer 1,2, Charles L. Barnes3, Samuel P. Hernández-Rivera4, and Jorge L. Ríos-Steiner 5 1Department of Physics, University of Massachusetts, Amherst, Massachusetts 01003, United States, e-mail: [email protected], [email protected] 2Department of Physics, University of Puerto Rico, Mayagüez, Puerto Rico 00681-9000, United States, e-mail: [email protected], [email protected], phone: +1 787 228 1380. 3Department of Chemistry, University of Missouri, Columbia, Missouri 65211, United States. 4ALERT-II DHS Center of Excellence for Explosives Research, Department of Chemistry, University of Puerto Rico, Mayagüez, Puerto Rico, 00681-9000, United States. 5Laboratory of Crystallography and Synthesis of New Materials, Department of Chemistry, University of Puerto Rico, Mayagüez, Puerto Rico, 00681-9000 United States, e-mail: [email protected], phone: +1 787 832 4040 ext 2538, Fax: +1 787 265 3849. January 15, 2019 Abstract The orthorhombic crystal structure of the novel molecular-inorganic polymer K2SeO4 · H2SeO3 was elucidated using single-crystal X-ray diffraction with MoKα radiation (λ= 0.71073 Å), performed at 100 and 298.15 K. The reported data is at 100 K since there were no struc- tural differences as compared to the room temperature. The technique revealed K2SeO4 ·H2SeO3 crystals to have space group P bcm with unit cell dimensions a = 8.8672(17) Å, b = 7.3355(14) Å, c = 11.999(2) Å and Z = 4. The unit cell volume obtained was V = 780.5(3) Å3 3 arXiv:1901.03765v1 [cond-mat.mtrl-sci] 11 Jan 2019 with a calculated density Dc = 2:980Mg=m . -

(12) United States Patent (10) Patent No.: US 9,018,217 B2 Ritzen Et Al
US009018217B2 (12) United States Patent (10) Patent No.: US 9,018,217 B2 Ritzen et al. (45) Date of Patent: Apr. 28, 2015 (54) PHENY LIMIDAZOLE DERIVATIVES AS 8,841,297 B2 9/2014 Ritzen et al. PDE10A ENZYME INHIBITORS 2008/0090891 A1 4/2008 Zelle 2012/0135987 A1 5, 2012 Ritzen et al. (71) Applicant: H. Lundbeck A/S, Valby-Copenhagen (DK) FOREIGN PATENT DOCUMENTS TW 2007/036246 A 10/2007 (72) Inventors: Andreas Ritzen, Copenhagen V. (DK); WO 2004/005290 A1 1, 2004 Morten Langgard, Glostrup (DK); Jan WO 2005/003129 A1 1, 2005 Kehler, Lyngby (DK); Jacob Nielsen, WO 2005/082883 A2 9, 2005 WO 2006/070284 A1 T 2006 Copenhagen V. (DK); John Paul WO 2007/077490 A2 7/2007 Kilburn, Haslev (DK); Mohamed M. WO 2007/098169 A1 8, 2007 Farah, Birmingham (GB) WO 2008.001182 A1 1, 2008 WO 2009/023179 A2 2, 2009 (73) Assignee: H. Lundbeck A/S, Valby (DK) WO 2010, 145668 12/2010 (*) Notice: Subject to any disclaimer, the term of this OTHER PUBLICATIONS patent is extended or adjusted under 35 Taiwan Examination report issued Jan. 13, 2014 in TW Application U.S.C. 154(b) by 0 days. No. 0981 19876 filed Jun. 15, 2009. International Search Report and Written Opinion issued Jul. 27, 2010 (21) Appl. No.: 14/488,554 for International Application No. PCT? DK2010/050 147 filed Jun. 17, 2010. (22) Filed: Sep. 17, 2014 Geyer et al., 2002, "Animal Models Relevant to Schizophrenia Dis orders'. Neuropsychopharmacology: The Fifth Generation of (65) Prior Publication Data Progress, pp. -

WO 2016/196440 Al 8 December 2016 (08.12.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/196440 Al 8 December 2016 (08.12.2016) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61P 3/04 (2006.01) A61K 33/40 (2006.01) kind of national protection available): AE, AG, AL, AM, A61P 9/10 (2006.01) A61K 38/44 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 35/74 (2015.01) A61K 31/17 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/US20 16/034973 KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (22) International Filing Date: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 3 1 May 2016 (3 1.05.2016) PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 62/169,480 1 June 2015 (01 .06.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/327,283 25 April 2016 (25.04.2016) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: XENO BIOSCIENCES INC. -

Preparation of Monodisperse Se Colloid Spheres and Se Nanowires
Nano Res (2008) 1: 403 411 DOI 10.1007/s12274-008-8040-5 00403 Research Article Preparation of Monodisperse Se Colloid Spheres and Se Nanowires Using Na2SeSO3 as Precursor Liping Liu, Qing Peng, and Yadong Li( ) Department of Chemistry, Tsinghua University, Beijing 100084, China Received: 3 September 2008 / Revised: 19 September 2008 /Accepted: 19 September 2008 ©Tsinghua Press and Springer-Verlag 2008. This article is published with open access at Springerlink.com ABSTRACT Nearly monodisperse spherical amorphous Se colloids are prepared by the dismutation of Na2SeSO3 solution at room temperature; by altering the pH of the solution, amorphous Se colloid spheres with sizes of about 120 nm, 200 nm, 300 nm, and 1 μm can be obtained. Se@Ag2Se core/shell spheres are successfully synthesized by using the obtained amorphous Se (a-Se) spheres as templates, indicating the potential applications of these Se nanomaterials in serving as soft templates for other selenides. Meanwhile, selenium nanowires are obtained through a “solid-solution-solid” growth process by dispersing the prepared Se spheres in ethanol. This simple and environmentally benign approach may offer more opportunities in the synthesis and applications of nanocrystal materials. KEYWORDS Na2SeSO3, dismutation, amorphous Se (a-Se) spheres, trigonal Se (t-Se) nanowires Introduction structures, as it is easily removed [4, 5]. Furthermore, selenium is a relatively active material and can react Materials which are composed of the element selenium with a variety of chemical reagents to form other play important roles in the fields of photonics materials, such as Ag Se, and CdSe [1, 6, 7], which and electronics due to its special photoconductive 2 possess wide potential applications as soft templates properties [1, 2]; the electrical conductivity of selenium for functional selenides. -

(Ii) 3,870,614 Underwood (45) Mar
United States Patent 19 (ii) 3,870,614 Underwood (45) Mar. 11, 1975 54 SELENIUM DEPOSITION Primary Examiner-Howard S. Williams 75 Inventor: John Duckles Underwood, Bishop Attorney, Agent, or Firm-John T. O'Halloran; Stortford, England Menotti J. Lombardi, Jr.; Vincent Ingrassia 73) Assignee: International Standard Electric Corporation, New York, N.Y. (22 Filed: June 6, 1974 57 ABSTRACT 21 Appl. No.: 476,986 This relates to a method of depositing selenium in a suitable form for the manufacture of rectifiers. Sele (52) U.S. Cl. ................................................ 204181 nium is electrophoretically deposited from a mixture (51) int. Cl............................................... B01k 5/02 of two selenium sols. The first sol is prepared by 58) Field of Search..................................... 204/18 chemical reduction of selenous acid and the second by grinding and colloid milling metallic selenium. 56 References Cited UNITED STATES PATENTS 8 Claims, 1 Drawing Figure 3,745,098 7, 1973 Brown et al........................ 2041 181 GREY SELENUM HEAT TREATMENT RECYCLE GRND AND COLLOD MLL WASH AND DECANT DSPERSE IN SO MEDUM ELECTROPHORESS TENTED KAR 1975 3,87O64. GREY SELENUM HEAT TREATMENT RECYCLE GRND AND COLLOD MLL WASH AND DECANT OSPERSE IN SOL MEDUM ELECTROPHORESS 3,870,614 1. 2 SELENIUM DEPOSITION After milling several times the colloidal particles are allowed to settle, the liquid is decanted and the parti BACKGROUND OF THE INVENTION cles are washed by decantation with an alcohol such as This invention relates to the electrophoretic deposi ethanol. The colloidal particles are then formed into a tion of selenium and in particular to a method of depos sol by dispersion in a sol medium typically consisting iting selenium in a form suitable for manufacturing rec of: tifiers.