Endothelin-Receptor Antagonists Are Proapoptotic and Antiproliferative in Human Colon Cancer Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Endothelin System and Therapeutic Application of Endothelin Receptor
xperim ACCESS Freely available online & E en OPEN l ta a l ic P in h l a C r m f o a c l a o n l o r g u y o J Journal of ISSN: 2161-1459 Clinical & Experimental Pharmacology Research Article Endothelin System and Therapeutic Application of Endothelin Receptor Antagonists Abebe Basazn Mekuria, Zemene Demelash Kifle*, Mohammedbrhan Abdelwuhab Department of Pharmacology, School of Pharmacy, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia ABSTRACT Endothelin is a 21 amino acid molecule endogenous potent vasoconstrictor peptide. Endothelin is synthesized in vascular endothelial and smooth muscle cells, as well as in neural, renal, pulmonic, and inflammatory cells. It acts through a seven transmembrane endothelin receptor A (ETA) and endothelin receptor B (ETB) receptors belongs to G protein-coupled rhodopsin-type receptor superfamily. This peptide involved in pathogenesis of cardiovascular disorder like (heart failure, arterial hypertension, myocardial infraction and atherosclerosis), renal failure, pulmonary arterial hypertension and it also involved in pathogenesis of cancer. Potentially endothelin receptor antagonist helps the treatment of the above disorder. Currently, there are a lot of trails both per-clinical and clinical on endothelin antagonist for various cardiovascular, pulmonary and cancer disorder. Some are approved by FAD for the treatment. These agents are including both selective and non-selective endothelin receptor antagonist (ETA/B). Currently, Bosentan, Ambrisentan, and Macitentan approved -

Self-Nano-Emulsifying Drug-Delivery Systems: from the Development to the Current Applications and Challenges in Oral Drug Delivery
pharmaceutics Review Self-Nano-Emulsifying Drug-Delivery Systems: From the Development to the Current Applications and Challenges in Oral Drug Delivery Aristote B. Buya 1,2 , Ana Beloqui 1, Patrick B. Memvanga 2 and Véronique Préat 1,* 1 Advanced Drug Delivery and Biomaterials, Louvain Drug Research Institute, Université Catholique de Louvain, Avenue Mounier 73, B1.73.12, 1200 Brussels, Belgium; [email protected] (A.B.B.); [email protected] (A.B.) 2 Pharmaceutics and Phytopharmaceutical Drug Development Research Group, Faculty of Pharmaceutical Sciences, University of Kinshasa, Kinshasa XI BP 212, Democratic Republic of the Congo; [email protected] * Correspondence: [email protected]; Tel.: +32-27647309 Received: 3 November 2020; Accepted: 5 December 2020; Published: 9 December 2020 Abstract: Approximately one third of newly discovered drug molecules show insufficient water solubility and therefore low oral bio-availability. Self-nano-emulsifying drug-delivery systems (SNEDDSs) are one of the emerging strategies developed to tackle the issues associated with their oral delivery. SNEDDSs are composed of an oil phase, surfactant, and cosurfactant or cosolvent. SNEDDSs characteristics, their ability to dissolve a drug, and in vivo considerations are determinant factors in the choice of SNEDDSs excipients. A SNEDDS formulation can be optimized through phase diagram approach or statistical design of experiments. The characterization of SNEDDSs includes multiple orthogonal methods required to fully control SNEDDS manufacture, stability, and biological fate. Encapsulating a drug in SNEDDSs can lead to increased solubilization, stability in the gastro-intestinal tract, and absorption, resulting in enhanced bio-availability. The transformation of liquid SNEDDSs into solid dosage forms has been shown to increase the stability and patient compliance. -

Comparison of Pharmacological Activity of Macitentan and Bosentan in Preclinical Models of Systemic and Pulmonary Hypertension
LFS-13929; No of Pages 7 Life Sciences xxx (2014) xxx–xxx Contents lists available at ScienceDirect Life Sciences journal homepage: www.elsevier.com/locate/lifescie Comparison of pharmacological activity of macitentan and bosentan in preclinical models of systemic and pulmonary hypertension Marc Iglarz ⁎, Alexandre Bossu, Daniel Wanner, Céline Bortolamiol, Markus Rey, Patrick Hess, Martine Clozel Drug Discovery Department, Actelion Pharmaceuticals Ltd, Gewerbestrasse 16, 4123 Allschwil, Switzerland article info abstract Article history: Aims: The endothelin (ET) system is a tissular system, as the production of ET isoforms is mostly autocrine or Received 29 October 2013 paracrine. Macitentan is a novel dual ETA/ETB receptor antagonist with enhanced tissue distribution and Accepted 12 February 2014 sustained receptor binding properties designed to achieve a more efficacious ET receptor blockade. To determine Available online xxxx if these features translate into improved efficacy in vivo, a study was designed in which rats with either systemic or pulmonary hypertension and equipped with telemetry were given macitentan on top of maximally effective Keywords: doses of another dual ET /ET receptor antagonist, bosentan, which does not display sustained receptor occupan- Endothelin A B Pharmacology cy and shows less tissue distribution. – Blood pressure Main methods: After establishing dose response curves of both compounds in conscious, hypertensive Dahl salt- Pulmonary hypertension sensitive and pulmonary hypertensive bleomycin-treated rats, macitentan was administered on top of the max- Rat imal effective dose of bosentan. Key findings: In hypertensive rats, macitentan 30 mg/kg further decreased mean arterial blood pressure (MAP) by 19 mm Hg when given on top of bosentan 100 mg/kg (n =9,p b 0.01 vs. -
Download Leaflet View the Patient Leaflet in PDF Format
long as you are taking Bosentan. We suggest you write the date of your most recent test and also of your next test (ask your doctor for the date) on the Patient Alert Package leaflet: Information for the user Card, to help you remember when your next test is due. Bosentan 62.5 mg film-coated tablets Blood tests for liver function These will be done every month for the duration of Bosentan 125 mg film-coated tablets treatment with Bosentan. After an increase in dose an additional test will be done after 2 weeks. bosentan Blood tests for anaemia Read all of this leaflet carefully before you start These will be done every month for the first 4 months taking this medicine because it contains important of treatment, then every 3 months after that, as patients information for you. taking Bosentan may get anaemia. - Keep this leaflet. You may need to read it again. If these results are abnormal, your doctor may decide - If you have any further questions, ask your doctor to reduce your dose or stop treatment with Bosentan or pharmacist. and to perform further tests to investigate the cause. - This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if Children and adolescents their signs of illness are the same as yours. Bosentan is not recommended in paediatric patients - If you get any side effects, talk to your doctor or with systemic sclerosis and ongoing digital ulcer pharmacist. This includes any possible side effects disease. Please see also section 3. -
![TRACLEER® [Bosentan] 62.5 Mg and 125 Mg Film-Coated Tablets](https://docslib.b-cdn.net/cover/9612/tracleer%C2%AE-bosentan-62-5-mg-and-125-mg-film-coated-tablets-1209612.webp)
TRACLEER® [Bosentan] 62.5 Mg and 125 Mg Film-Coated Tablets
TRACLEER® [bosentan] 62.5 mg and 125 mg film-coated tablets Use of TRACLEER® requires attention to two significant concerns: 1) potential for serious liver injury, and 2) potential damage to a fetus. WARNING: Potential liver injury TRACLEER® causes at least 3-fold (upper limit of normal; ULN) elevation of liver aminotransferases (ALT and AST) in about 11% of patients, accompanied by elevated bilirubin in a small number of cases. Because these changes are a marker for potential serious liver injury, serum aminotransferase levels must be measured prior to initiation of treatment and then monthly (see WARNINGS: Potential Liver Injury and DOSAGE AND ADMINISTRATION). To date, in a setting of close monitoring, elevations have been reversible, within a few days to 9 weeks, either spontaneously or after dose reduction or discontinuation, and without sequelae. Elevations in aminotransferases require close attention (see DOSAGE AND ADMINISTRATION). TRACLEER® should generally be avoided in patients with elevated aminotransferases (> 3 x ULN) at baseline because monitoring liver injury may be more difficult. If liver aminotransferase elevations are accompanied by clinical symptoms of liver injury (such as nausea, vomiting, fever, abdominal pain, jaundice, or unusual lethargy or fatigue) or increases in bilirubin ≥ 2 x ULN, treatment should be stopped. There is no experience with the re-introduction of TRACLEER® in these circumstances. CONTRAINDICATION: Pregnancy TRACLEER® (bosentan) is very likely to produce major birth defects if used by pregnant women, as this effect has been seen consistently when it is administered to animals (see CONTRAINDICATIONS). Therefore, pregnancy must be excluded before the start of treatment with TRACLEER® and prevented thereafter by the use of a reliable method of contraception. -

84 November 2008 Produced by NHS Tayside Drug and Therapeutics Committee Medicines Advisory Group (MAG)
TAYSIDE PRESCRIBER Tayside DTC Supplement No 84 November 2008 Produced by NHS Tayside Drug and Therapeutics Committee Medicines Advisory Group (MAG) SMC Advice issued in November 2008 Medicine Indication Local recommendation Comments and useful category links Ambrisentan tablets Treatment of patients with class Restricted for use within SMC advice (Volibris®) II and III pulmonary arterial national treatment centre SPC link hypertension to improve exercise capacity Anidulafungin powder and Treatment of invasive Pending AMG decision SMC advice solvent (Ecalta®) - candidiasis in adult non- SPC link Resubmission neutropenic patients Bosentan film-coated tablets Class II pulmonary arterial Not recommended SMC advice (Tracleer®) - hypertension Bosentan was previously Non submission accepted in 2003 for restricted use in grade III pulmonary arterial hypertension Icatibant solution for Symptomatic treatment of acute Not recommended SMC advice subcutaneous injection attacks of hereditary (Firazyr®) angioedema in adults Insulin glulisine solution for Treatment of adolescents and Non-formulary SMC advice subcutaneous injection children, 6 years or older with SPC link (Apidra®) - Abbreviated diabetes mellitus where a short Diabetes Handbook submission acting insulin analogue is appropriate Insulin lispro solution for Adults and children with Non-formulary SMC advice injection (Humalog® diabetes mellitus SPC link KwikPen) - Diabetes Handbook Abbreviated submission Insulin lispro (Humalog® Patients with diabetes mellitus, Non-formulary SMC advice Mix25 -

"Tracleer, INN- Bosentan"
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Tracleer 62.5 mg film-coated tablets Tracleer 125 mg film-coated tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Tracleer 62.5 mg film-coated tablets Each film-coated tablet contains 62.5 mg bosentan (as monohydrate). Tracleer 125 mg film-coated tablets Each film-coated tablet contains 125 mg bosentan (as monohydrate). Excipient with known effect This medicine contains less than 1 mmol sodium (23 mg) per tablet, that is to say essentially ‘sodium-free’. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Film-coated tablet (tablets): Tracleer 62.5 mg film-coated tablets Orange-white, round, biconvex, film-coated tablets, embossed with “62,5” on one side. Tracleer 125 mg film-coated tablets Orange-white, oval, biconvex, film-coated tablets, embossed with “125” on one side. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Treatment of pulmonary arterial hypertension (PAH) to improve exercise capacity and symptoms in patients with WHO functional class III. Efficacy has been shown in: Primary (idiopathic and heritable) pulmonary arterial hypertension Pulmonary arterial hypertension secondary to scleroderma without significant interstitial pulmonary disease Pulmonary arterial hypertension associated with congenital systemic-to-pulmonary shunts and Eisenmenger’s physiology Some improvements have also been shown in patients with pulmonary arterial hypertension WHO functional class II (see section 5.1). Tracleer is also indicated to reduce the number of new digital ulcers in patients with systemic sclerosis and ongoing digital ulcer disease (see section 5.1). 2 4.2 Posology and method of administration Method of administration Tablets are to be taken orally morning and evening, with or without food. -

Advanced Control Specialty Formulary® Is a Guide Within Select Therapeutic Categories for Clients, Plan Members and Health Care Providers
October 2021 ® Advanced Control Specialty Formulary The CVS Caremark® Advanced Control Specialty Formulary® is a guide within select therapeutic categories for clients, plan members and health care providers. Generics should be considered the first line of prescribing. If there is no generic available, there may be more than one brand-name medicine to treat a condition. These preferred brand-name medicines are listed to help identify products that are clinically appropriate and cost-effective. Generics listed in therapeutic categories are for representational purposes only. This is not an all-inclusive list. This list represents brand products in CAPS, branded generics in upper- and lowercase Italics, and generic products in lowercase italics. PLAN MEMBER HEALTH CARE PROVIDER Your benefit plan provides you with a prescription benefit program Your patient is covered under a prescription benefit plan administered administered by CVS Caremark. Ask your doctor to consider by CVS Caremark. As a way to help manage health care costs, prescribing, when medically appropriate, a preferred medicine from authorize generic substitution whenever possible. If you believe a this list. Take this list along when you or a covered family member brand-name product is necessary, consider prescribing a brand name sees a doctor. on this list. Please note: Please note: • Your specific prescription benefit plan design may not cover • Generics should be considered the first line of prescribing. certain products or categories, regardless of their appearance in • The member's prescription benefit plan design may alter coverage this document. Products recently approved by the U.S. Food and of certain products or vary copay amounts based on the condition Drug Administration (FDA) may not be covered upon release being treated. -

List of Formulary Drug Removals
July 2021 Formulary Drug Removals Below is a list of medicines by drug class that have been removed from your plan’s formulary. If you continue using one of the drugs listed below and identified as a Formulary Drug Removal, you may be required to pay the full cost. If you are currently using one of the formulary drug removals, ask your doctor to choose one of the generic or brand formulary options listed below. Category Formulary Drug Formulary Options Drug Class Removals Acromegaly SANDOSTATIN LAR SOMATULINE DEPOT SIGNIFOR LAR SOMAVERT Allergies dexchlorpheniramine levocetirizine Antihistamines Diphen Elixir RyClora CARBINOXAMINE TABLET 6 MG Allergies BECONASE AQ flunisolide spray, fluticasone spray, mometasone spray, DYMISTA Nasal Steroids / Combinations OMNARIS QNASL ZETONNA Anticonvulsants topiramate ext-rel capsule carbamazepine, carbamazepine ext-rel, clobazam, divalproex sodium, (generics for QUDEXY XR only) divalproex sodium ext-rel, gabapentin, lamotrigine, lamotrigine ext-rel, levetiracetam, levetiracetam ext-rel, oxcarbazepine, phenobarbital, phenytoin, phenytoin sodium extended, primidone, rufinamide, tiagabine, topiramate, valproic acid, zonisamide, FYCOMPA, OXTELLAR XR, TROKENDI XR, VIMPAT, XCOPRI BANZEL SUSPENSION clobazam, lamotrigine, rufinamide, topiramate, TROKENDI XR ONFI SABRIL vigabatrin ZONEGRAN carbamazepine, carbamazepine ext-rel, divalproex sodium, divalproex sodium ext-rel, gabapentin, lamotrigine, lamotrigine ext-rel, levetiracetam, levetiracetam ext-rel, oxcarbazepine, phenobarbital, phenytoin, phenytoin sodium -

Pennsylvania Department of Human Services Statewide Preferred Drug List (PDL)*
Pennsylvania Department of Human Services Statewide Preferred Drug List (PDL)* *The Statewide PDL is not an all-inclusive list of drugs covered by Medical Assistance. Drugs in Statewide PDL classes that are new to market will be non-preferred until reviewed by the DHS Pharmacy and Therapeutics Committee. Fee-for-Service‡ (FFS) Pharmacy General Prior Authorization Requirements: https://dhs.pa.gov/providers/Pharmacy-Services/Pages/Pharmacy-Prior-Authorization-General-Requirements.aspx FFS† Pharmacy Prior Authorization Clinical Guidelines: https://www.dhs.pa.gov/providers/Pharmacy-Services/Pages/Clinical-Guidelines.aspx FFS‡ Pharmacy Prior Authorization Fax Forms: https://www.dhs.pa.gov/providers/Pharmacy-Services/Pages/Pharmacy-Services-Fax-Forms.aspx FFS‡ Pharmacy Quantity Limits/Daily Dose Limits: https://dhs.pa.gov/providers/Pharmacy-Services/Pages/Quantity-Limits-and-Daily-Dose-Limits.aspx ‡This information is specific to FFS. Please refer to each managed care organization’s (MCO) website for MCO prior authorization procedures, prior authorization fax request forms, and quantity limits. †Prior authorization guidelines for drugs and products included in the Statewide PDL apply to FFS and the Pennsylvania Medical Assistance MCOs. Prior authorization guidelines for drugs and products not included in the Statewide PDL are specific to FFS. Please refer to each MCO’s website for MCO-specific prior authorization requirements for drugs and products not included in the Statewide PDL. ACNE AGENTS, ORAL Preferred Agents Non-Preferred Agents Amnesteem -
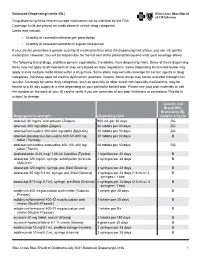
Balanced Drug List Dispensing Limits
Balanced Dispensing Limits (DL) Drug dispensing limits help encourage medication use as intended by the FDA. Coverage limits are placed on medications in certain drug categories. Limits may include: • Quantity of covered medication per prescription • Quantity of covered medication in a given time period If your doctor prescribes a greater quantity of medication than what the dispensing limit allows, you can still get the medication. However, you will be responsible for the full cost of the prescription beyond what your coverage allows. The following brand drugs, and their generic equivalents, if available, have dispensing limits. Some of these dispensing limits may not apply to all members or may vary based on state regulations. Some dispensing limits listed below may apply across multiple medications within a drug class. Some plans may exclude coverage for certain agents or drug categories, like those used for erectile dysfunction (example: Viagra). Some drugs may not be available through mail service. Coverage for some drug categories, such as specialty or other select non‑specialty medications, may be limited to a 30‑day supply at a time depending on your particular benefit plan. Please see your plan materials or call the number on the back of your ID card to verify if you are uncertain of any plan limitations or exclusions. This list is subject to change. Generic and Brand (BG), Brand Only (B), Drug (generic) strength Dispensing Limit Generic only (G) abacavir 20 mg/mL oral solution (Ziagen) 960 mL per 30 days BG abacavir 300 mg tablet -

Bosentan Or Macitentan Therapy in Chronic Thromboembolic Pulmonary Hypertension?
Lung (2019) 197:753–760 https://doi.org/10.1007/s00408-019-00274-9 PULMONARY HYPERTENSION Bosentan or Macitentan Therapy in Chronic Thromboembolic Pulmonary Hypertension? M. C. J. van Thor1,2 · L. ten Klooster2 · R. J. Snijder2 · J. C. Kelder3 · J. J. Mager2 · M. C. Post1 Received: 28 June 2019 / Accepted: 19 September 2019 / Published online: 3 October 2019 © Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Objective Research comparing bosentan and macitentan in chronic thromboembolic pulmonary hypertension (CTEPH) is scarce, although macitentan might have superior pharmacologic properties. We present the frst real-world, 2-year follow-up results and compare clinical outcomes of both drugs in CTEPH. Methods All consecutive, technical inoperable or residual CTEPH patients receiving bosentan or macitentan, diagnosed in our multidisciplinary team between January 2003 and January 2019, were included. We report and compare survival, clini- cal worsening (CW), adverse events, WHO FC, NT-proBNP and 6-min walking test (6MWT) until 2 years after medication initiation. Results In total, 112 patients receiving bosentan or macitentan (58% female, mean age 62 ± 14 years, 68% WHO FC III/IV, 51% bosentan) could be included. Mean treatment duration was 1.9 ± 0.4 years for bosentan and 1.2 ± 0.6 years for macitentan. Two-year survival rate was 91% for bosentan and 80% for macitentan (HR mortality macitentan 1.85 [0.56–6.10], p = 0.31). Two-year CW-free survival was 81% and 58%, respectively (HR CW macitentan 2.16 [0.962–4.87], p = 0.06). Right atrial pressure, cardiac output (for mortality alone) and 6MWT lowest saturation were multivariate predictors at baseline.