209627Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ANNOVERA™ (Segesterone Acetate and Ethinyl Estradiol Vaginal System) • Risk of Liver Enzyme Elevations with Concomitant Hepatitis C Initial U.S
HIGHLIGHTS OF PRESCRIBING INFORMATION ANNOVERA™ no earlier than 4 weeks after delivery, in females who These highlights do not include all the information needed to use are not breastfeeding. Consider cardiovascular risk factors before ANNOVERA™ safely and effectively. initiating in all females, particularly those over 35 years. (5.1, 5.5) See Full Prescribing Information for ANNOVERA™. • Liver Disease: Discontinue if jaundice occurs. (5.2) ANNOVERA™ (segesterone acetate and ethinyl estradiol vaginal system) • Risk of Liver Enzyme Elevations with Concomitant Hepatitis C Initial U.S. Approval: 2018 Treatment: Stop ANNOVERA™ prior to starting therapy with the combination drug regimen ombitasvir/paritaprevir/ritonavir. ANNOVERA™ can be restarted 2 weeks following completion of this WARNING: CIGARETTE SMOKING AND regimen. (5.3) SERIOUS CARDIOVASCULAR EVENTS • Hypertension: Do not prescribe ANNOVERA™ for females with See full prescribing information for complete boxed warning. uncontrolled hypertension or hypertension with vascular disease. If • Females over 35 years old who smoke should not use used in females with well-controlled hypertension, monitor blood ANNOVERA™. (4) pressure and stop use if blood pressure rises significantly. (5.4) • Cigarette smoking increases the risk of serious cardiovascular • Carbohydrate and lipid metabolic effects: Monitor glucose in pre events from combination hormonal contraceptive (CHC) use. (4) diabetic and diabetic females taking ANNOVERA™. Consider an alternate contraceptive method for females with uncontrolled ----------------------------INDICATIONS AND USAGE-------------------------- dyslipidemias. (5.7) ANNOVERA™ is a progestin/estrogen CHC indicated for use by females of • Headache: Evaluate significant change in headaches and discontinue reproductive potential to prevent pregnancy. (1) ANNOVERA™ if indicated. (5.8) Limitation of use: Not adequately evaluated in females with a body mass index • Bleeding Irregularities and Amenorrhea: May cause irregular bleeding of >29 kg/m2. -

209627Orig1s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 209627Orig1s000 MULTI-DISCIPLINE REVIEW Summary Review Office Director Cross Discipline Team Leader Review Clinical Review Non-Clinical Review Statistical Review Clinical Pharmacology Review Reviewers of Multi-Disciplinary Review and Evaluation SECTIONS OFFICE/ AUTHORED/ ACKNOWLEDGED/ DISCIPLINE REVIEWER DIVISION APPROVED Mark Seggel, Ph.D. OPQ/ONDP/DNDP2 Authored: Section 4.2 Digitally signed by Mark R. Seggel -S CMC Lead DN: c=US, o=U.S. Government, ou=HHS, ou=FDA, ou=People, cn=Mark R. Signature: Mark R. Seggel -S Seggel -S, 0.9.2342.19200300.100.1.1=1300071539 Date: 2018.08.08 16:29:15 -04'00' Frederic Moulin, DVM, PhD OND/ODE3/DBRUP Authored: Section 5 Pharmacology/ Digitally signed by Frederic Moulin -S Toxicology DN: c=US, o=U.S. Government, ou=HHS, ou=FDA, ou=People, Reviewer Signature: Frederic Moulin -S 0.9.2342.19200300.100.1.1=2001708658, cn=Frederic Moulin -S Date: 2018.08.08 15:26:57 -04'00' Kimberly Hatfield, PhD OND/ODE3/DBRUP Approved: Section 5 Pharmacology/ Toxicology Digitally signed by Kimberly P. Hatfield -S DN: c=US, o=U.S. Government, ou=HHS, ou=FDA, ou=People, Team Leader Signature: Kimberly P. Hatfield -S 0.9.2342.19200300.100.1.1=1300387215, cn=Kimberly P. Hatfield -S Date: 2018.08.08 14:56:10 -04'00' Li Li, Ph.D. OCP/DCP3 Authored: Sections 6 and 17.3 Clinical Pharmacology Dig ta ly signed by Li Li S DN c=US o=U S Government ou=HHS ou=FDA ou=People Reviewer cn=Li Li S Signature: Li Li -S 0 9 2342 19200300 100 1 1=20005 08577 Date 2018 08 08 15 39 23 04'00' Doanh Tran, Ph.D. -

Health Care Reform
Health Care Reform Preventative Drug list At Magellan Rx Management, we are driven to helping our clients most effectively manage changes in the prescription drug environment. As part of the Patient Protection and Affordable Care Act (PPACA), effective on or after August 1, 2012 non-grandfathered plans are required to cover select FDA-approved drug products related to preventive health services for Adults, Children and Women without a member having to pay a copayment, co-insurance, or meet a deductible. As your pharmacy benefits manager, we will support coverage of specific products at $0 copay as prescription benefit plans become subject to the law. Magellan Rx Management will also routinely update FDA-approved product lists to comply with the Preventive Health mandates and require prescriptions for product coverage. MRx has created ten specific lists that address the preventive health requirements above. A plan is required to cover all the drug lists below, either through the pharmacy or medical benefits. If the plan needs advice regarding coverage, MRx recommends the plan connect with their respective legal counsel. The content of the list will be maintained by Magellan Rx Management and will be continually reviewed and updated to ensure compliance with healthcare reform mandates. UPDATE: For plan years beginning on or after September 24, 2014 (January 1, 2015 for calendar year plans), non-grandfathered health plans are required to cover prescription medications designed to reduce the risk of breast cancer in women, without cost-sharing, subject to reasonable medical management. These required drugs have been added to the “Preventive Health for Adults, Children and Women: Prescription and OTC Products” list below. -

Vaginal Administration of Contraceptives
Scientia Pharmaceutica Review Vaginal Administration of Contraceptives Esmat Jalalvandi 1,*, Hafez Jafari 2 , Christiani A. Amorim 3 , Denise Freitas Siqueira Petri 4 , Lei Nie 5,* and Amin Shavandi 2,* 1 School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK 2 BioMatter Unit, École Polytechnique de Bruxelles, Université Libre de Bruxelles, Avenue F.D. Roosevelt, 50-CP 165/61, 1050 Brussels, Belgium; [email protected] 3 Pôle de Recherche en Gynécologie, Institut de Recherche Expérimentale et Clinique, Université Catholique de Louvain, 1200 Brussels, Belgium; [email protected] 4 Fundamental Chemistry Department, Institute of Chemistry, University of São Paulo, Av. Prof. Lineu Prestes 748, São Paulo 05508-000, Brazil; [email protected] 5 College of Life Sciences, Xinyang Normal University, Xinyang 464000, China * Correspondence: [email protected] (E.J.); [email protected] (L.N.); [email protected] (A.S.); Tel.: +32-2-650-3681 (A.S.) Abstract: While contraceptive drugs have enabled many people to decide when they want to have a baby, more than 100 million unintended pregnancies each year in the world may indicate the contraceptive requirement of many people has not been well addressed yet. The vagina is a well- established and practical route for the delivery of various pharmacological molecules, including contraceptives. This review aims to present an overview of different contraceptive methods focusing on the vaginal route of delivery for contraceptives, including current developments, discussing the potentials and limitations of the modern methods, designs, and how well each method performs for delivering the contraceptives and preventing pregnancy. -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

2021 Formulary List of Covered Prescription Drugs
2021 Formulary List of covered prescription drugs This drug list applies to all Individual HMO products and the following Small Group HMO products: Sharp Platinum 90 Performance HMO, Sharp Platinum 90 Performance HMO AI-AN, Sharp Platinum 90 Premier HMO, Sharp Platinum 90 Premier HMO AI-AN, Sharp Gold 80 Performance HMO, Sharp Gold 80 Performance HMO AI-AN, Sharp Gold 80 Premier HMO, Sharp Gold 80 Premier HMO AI-AN, Sharp Silver 70 Performance HMO, Sharp Silver 70 Performance HMO AI-AN, Sharp Silver 70 Premier HMO, Sharp Silver 70 Premier HMO AI-AN, Sharp Silver 73 Performance HMO, Sharp Silver 73 Premier HMO, Sharp Silver 87 Performance HMO, Sharp Silver 87 Premier HMO, Sharp Silver 94 Performance HMO, Sharp Silver 94 Premier HMO, Sharp Bronze 60 Performance HMO, Sharp Bronze 60 Performance HMO AI-AN, Sharp Bronze 60 Premier HDHP HMO, Sharp Bronze 60 Premier HDHP HMO AI-AN, Sharp Minimum Coverage Performance HMO, Sharp $0 Cost Share Performance HMO AI-AN, Sharp $0 Cost Share Premier HMO AI-AN, Sharp Silver 70 Off Exchange Performance HMO, Sharp Silver 70 Off Exchange Premier HMO, Sharp Performance Platinum 90 HMO 0/15 + Child Dental, Sharp Premier Platinum 90 HMO 0/20 + Child Dental, Sharp Performance Gold 80 HMO 350 /25 + Child Dental, Sharp Premier Gold 80 HMO 250/35 + Child Dental, Sharp Performance Silver 70 HMO 2250/50 + Child Dental, Sharp Premier Silver 70 HMO 2250/55 + Child Dental, Sharp Premier Silver 70 HDHP HMO 2500/20% + Child Dental, Sharp Performance Bronze 60 HMO 6300/65 + Child Dental, Sharp Premier Bronze 60 HDHP HMO -

Contraceptive Update: New Methods, New Guidelines
UCSF Essentials of Primary Care Conference Squaw Creek, CA August 8, 2019 Contraceptive Update: New Methods, New Guidelines Michael S. Policar, MD, MPH Professor Emeritus of Ob, Gyn, and Repro Sci UCSF School of Medicine [email protected] A. Every day (or moreDo You often) Use the US Medical Eligibility B. Occasionally (a few times a week) C. Rarely (a few times a month)Criteria (MEC) in Your Practice? D. Never…they don’t apply to my E. I’ve neverpractice heard of them! • Bayer: litigation consultant • Disclosures relevant to this talk Sebela Pharmaceuticals: – Investigator trainer and proctor in phase III trial of a copper IUD (VeraCept) 6% 50% 10% 17% Every day (or more often) 17% Occasionally (a few times a ... Rarely (a few times a month) Never…they don’t apply to .. I’ve never heard of them! MMWR. July 29, 2016 65 (3):1-103 On-line at: https://www.cdc.gov/mmwr/volumes/65/rr/pdfs/rr6503.pdf US Medical Eligibility Criteria Cat Definition Recommendation 1 No restriction in use Use the method 2 Advantages generally More than usual follow-up outweigh theoretical or needed proven risks 3 Theoretical or proven risks Clinical judgment that the outweigh advantages patient can use safely 4 Unacceptable health risk if the Do not use the method method is used • U.S. Selected Practice Recommendations for Contraceptive Use, 2016. MMWR July 29, 2016. 65(4);1–66 2016 Updates to the US MEC and SPR • New recommendations for women with – Cystic fibrosis – Multiple sclerosis • Interactions with SSRIs and St. -

Preferred Drug List
Kansas State Employee ANALGESICS Second Generation cefprozil Health Plan NSAIDs cefuroxime axetil diclofenac sodium delayed-rel Preferred Drug List diflunisal Third Generation etodolac cefdinir 2021 ibuprofen cefixime (SUPRAX) meloxicam nabumetone Erythromycins/Macrolides naproxen sodium tabs azithromycin naproxen tabs clarithromycin oxaprozin clarithromycin ext-rel sulindac erythromycin delayed-rel erythromycin ethylsuccinate NSAIDs, COMBINATIONS erythromycin stearate diclofenac sodium delayed-rel/misoprostol fidaxomicin (DIFICID) Effective 04/01/2021 NSAIDs, TOPICAL Fluoroquinolones diclofenac sodium gel 1% ciprofloxacin For questions or additional information, diclofenac sodium soln levofloxacin access the State of Kansas website at moxifloxacin http://www.kdheks.gov/hcf/sehp or call COX-2 INHIBITORS Penicillins the Kansas State Employees Prescription celecoxib amoxicillin Drug Program at 1-800-294-6324. amoxicillin/clavulanate The Preferred Drug List is subject to change. GOUT amoxicillin/clavulanate ext-rel To locate covered prescriptions online, allopurinol ampicillin access the State of Kansas website at colchicine tabs dicloxacillin http://www.kdheks.gov/hcf/sehp for the probenecid penicillin VK most current drug list. colchicine (MITIGARE) Tetracyclines What is a Preferred Drug List? OPIOID ANALGESICS doxycycline hyclate A Preferred Drug List is a list of safe and buprenorphine transdermal minocycline cost-effective drugs, chosen by a committee codeine/acetaminophen tetracycline of physicians and pharmacists. Drug lists fentanyl -

Wednesday, June 12, 2019 4:00Pm
Wednesday, June 12, 2019 4:00pm Oklahoma Health Care Authority 4345 N. Lincoln Blvd. Oklahoma City, OK 73105 The University of Oklahoma Health Sciences Center COLLEGE OF PHARMACY PHARMACY MANAGEMENT CONSULTANTS MEMORANDUM TO: Drug Utilization Review (DUR) Board Members FROM: Melissa Abbott, Pharm.D. SUBJECT: Packet Contents for DUR Board Meeting – June 12, 2019 DATE: June 5, 2019 Note: The DUR Board will meet at 4:00pm. The meeting will be held at 4345 N. Lincoln Blvd. Enclosed are the following items related to the June meeting. Material is arranged in order of the agenda. Call to Order Public Comment Forum Action Item – Approval of DUR Board Meeting Minutes – Appendix A Update on Medication Coverage Authorization Unit/Use of Angiotensin Converting Enzyme Inhibitor (ACEI)/ Angiotensin Receptor Blocker (ARB) Therapy in Patients with Diabetes and Hypertension (HTN) Mailing Update – Appendix B Action Item – Vote to Prior Authorize Aldurazyme® (Laronidase) and Naglazyme® (Galsulfase) – Appendix C Action Item – Vote to Prior Authorize Plenvu® [Polyethylene Glycol (PEG)-3350/Sodium Ascorbate/Sodium Sulfate/Ascorbic Acid/Sodium Chloride/Potassium Chloride] – Appendix D Action Item – Vote to Prior Authorize Consensi® (Amlodipine/Celecoxib) and Kapspargo™ Sprinkle [Metoprolol Succinate Extended-Release (ER)] – Appendix E Action Item – Vote to Update the Prior Authorization Criteria For H.P. Acthar® Gel (Repository Corticotropin Injection) – Appendix F Action Item – Vote to Prior Authorize Fulphila® (Pegfilgrastim-jmdb), Nivestym™ (Filgrastim-aafi), -

Preventive Care Services: Contraception
Preventive Care Services: Contraception Preventive Care Coverage at No Cost to You Effective Jan. 1, 2021 Your health plan may provide certain contraceptive coverage as a benefit of membership, at no cost to you when you use a pharmacy or doctor in your health plan's network. There is no copay, deductible or coinsurance, even if your deductible or out-of-pocket maximum has not been met. Coverage for contraceptives can vary depending on the type of plan you are enrolled in, as well as your prescription drug list. If you are using a contraceptive not listed under the Contraceptive Product Coverage, then copays, coinsurance or deductible may apply. Check your drug list or call the number listed on your member ID card to find out what products are covered at no cost share under your plan. Contraception* The following contraceptive items and services may be covered under the medical or pharmacy benefit without cost-sharing when provided by a pharmacy or doctor in your health plan's network. This list is not all inclusive. Additional products may be covered at no additional cost. • One or more prescribed products within each of the categories approved by the FDA for use as a method of contraception • FDA-approved contraceptives available over the counter (i.e. foam, sponge, female condoms), when prescribed by a physician • The morning after pill • Injections such as IM DEPO-PROVERA and DEPO-SUBQ PROVERA 104 may be covered under the medical or pharmacy benefit • Medical devices such as diaphragms, cervical caps and contraceptive implants may -
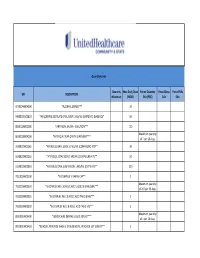
UHC PA Community and State Quantity Limit List
Quantity Limits Quantity Max Daily Dose Period Quantity Period Days Period Fills GPI DESCRIPTION Maximum (MDD) Edit (PQE) Edit Edit 97703040004300 *ALCOHOL SWABS*** 10 34000003101810 *AMLODIPINE BESYLATE ORAL SUSP 1 MG/ML (CMPD KIT) (BASE EQ)* 10 88501000002000 *ARTIFICIAL SALIVA - SOLUTION*** 120 Maximum quantity 86202000004200 *ARTIFICIAL TEAR OPHTH OINTMENT*** of 7 per 26 days. 33200020002065 *ATENOLOL ORAL SOLN 10 MG/ML (COMPOUND KIT)** 20 33200020002055 *ATENOLOL ORAL SOLN 2 MG/ML (COMPOUND KIT)** 50 33200020001810 *ATENOLOL ORAL SUSPENSION 1 MG/ML (CMPD KIT)** 200 78110000000100 *B-COMPLEX VITAMIN CAP** 1 Maximum quantity 78133000000920 *B-COMPLEX W/ C & FOLIC ACID LIQUID 0.9 MG/5ML*** of 237 per 26 days. 78133000000325 *B-COMPLEX W/ C & FOLIC ACID TAB 0.8 MG*** 1 78133000000330 *B-COMPLEX W/ C & FOLIC ACID TAB 1 MG*** 1 Maximum quantity 88350010006400 *BENZOCAINE DENTAL LIQUID 20% KIT*** of 1 per 26 days. 90050010006410 *BENZOYL PEROXIDE WASH 2.5% & BENZOYL PEROXIDE LOT 10% KIT** 1 47300005100100 *BIFIDOBACTERIUM BIFIDUM CAP** 2 97202007100900 *BLOOD GLUCOSE CALIBRATION - LIQUID*** 0.04 Maximum quantity 97202011006200 *BLOOD GLUCOSE METER DISPOSABLE DEVICE WITH TEST STRIPS*** of 1 per 999 days. Maximum quantity 97202010006200 *BLOOD GLUCOSE MONITORING DEVICES*** of 1 per 365 days. Maximum quantity 97202010006410 *BLOOD GLUCOSE MONITORING KIT W/ DEVICE*** of 1 per 365 days. Maximum quantity 97202010006400 *BLOOD GLUCOSE MONITORING KIT*** of 1 per 365 days. Maximum quantity 97750010006200 *BLOOD PRESSURE MONITORING - DEVICE*** of 1 per 999 days. Maximum quantity 9025990212B120 *CALCIPOTRIENE CR 0.005% & DIMETHICONE CR 5% THERAPY PACK*** of 1 per 26 days. 79109903450340 *CALCIUM CARB-VIT D W/ MINERALS TABS 600 MG-200 UNIT*** 5 79109903450355 *CALCIUM CARB-VIT D W/ MINERALS TABS 600 MG-800 UNIT*** 2 Maximum quantity 6610990420B120 *CELECOXIB CAP 200 MG & METH SAL-MEN-CAPSAICIN LIQD THER PK* of 1 per 26 days. -

Orange Book Cumulative Supplement 08 August 2019
CUMULATIVE SUPPLEMENT 8 AUGUST 2019 APPROVED DRUG PRODUCTS WITH THERAPEUTIC EQUIVALENCE EVALUATIONS 39th EDITION Department of Health and Human Services Food and Drug Administration Office of Medical Products and Tobacco Center for Drug Evaluation and Research Office of Generic Drugs Office of Generic Drug Policy 2019 Prepared By Food and Drug Administration Office of Medical Products and Tobacco Center for Drug Evaluation and Research Office of Generic Drugs Office of Generic Drug Policy APPROVED DRUG PRODUCTS with THERAPEUTIC EQUIVALENCE EVALUATIONS 39th EDITION Cumulative Supplement 8 August 2019 CONTENTS PAGE 1.0 INTRODUCTION ......................................................................................................................................... v 1.1 How to use the Cumulative Supplement ............................................................................................ v 1.2 Cumulative Supplement Content ...................................................................................................... vi 1.3 Applicant Name Changes ................................................................................................................ vii 1.4 Levothyroxine Sodium....................................................................................................................... ix 1.5 Availability of the Edition .................................................................................................................... x 1.6 Report of Counts for the Prescription Drug Product List ..................................................................