Fast Rhythmic Bursting Can Be Induced in Layer 2/3 Cortical Neurons by Enhancing Persistent Naϩ Conductance Or by Blocking BK Channels
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Interactions Among Diameter, Myelination, and the Na/K Pump Affect Axonal Resilience to High-Frequency Spiking
Interactions among diameter, myelination, and the Na/K pump affect axonal resilience to high-frequency spiking Yunliang Zanga,b and Eve Mardera,b,1 aVolen National Center for Complex Systems, Brandeis University, Waltham, MA 02454; and bDepartment of Biology, Brandeis University, Waltham, MA 02454 Contributed by Eve Marder, June 22, 2021 (sent for review March 25, 2021; reviewed by Carmen C. Canavier and Steven A. Prescott) Axons reliably conduct action potentials between neurons and/or (16–21). It remains unknown how these directly correlated pro- other targets. Axons have widely variable diameters and can be cesses, fast spiking and slow Na+ removal, interact in the control myelinated or unmyelinated. Although the effect of these factors of reliable spike generation and propagation. Furthermore, it is on propagation speed is well studied, how they constrain axonal unclear whether reducing Na+-andK+-current overlap can con- resilience to high-frequency spiking is incompletely understood. sistently decrease the rate of Na+ influx and accordingly enhance Maximal firing frequencies range from ∼1Hzto>300 Hz across axonal reliability to propagate high-frequency spikes. Conse- neurons, but the process by which Na/K pumps counteract Na+ quently, by modeling the process of Na+ removal in unmyelinated influx is slow, and the extent to which slow Na+ removal is com- and myelinated axons, we systematically explored the effects of patible with high-frequency spiking is unclear. Modeling the pro- diameter, myelination, and Na/K pump density on spike propagation cess of Na+ removal shows that large-diameter axons are more reliability. resilient to high-frequency spikes than are small-diameter axons, because of their slow Na+ accumulation. -

Energy Expenditure Computation of a Single Bursting Neuron
Cognitive Neurodynamics (2019) 13:75–87 https://doi.org/10.1007/s11571-018-9503-3 (0123456789().,-volV)(0123456789().,-volV) ORIGINAL ARTICLE Energy expenditure computation of a single bursting neuron 2 1,2 2 2 Fengyun Zhu • Rubin Wang • Xiaochuan Pan • Zhenyu Zhu Received: 17 October 2017 / Revised: 22 August 2018 / Accepted: 28 August 2018 / Published online: 3 September 2018 Ó The Author(s) 2018 Abstract Brief bursts of high-frequency spikes are a common firing pattern of neurons. The cellular mechanisms of bursting and its biological significance remain a matter of debate. Focusing on the energy aspect, this paper proposes a neural energy calculation method based on the Chay model of bursting. The flow of ions across the membrane of the bursting neuron with or without current stimulation and its power which contributes to the change of the transmembrane electrical potential energy are analyzed here in detail. We find that during the depolarization of spikes in bursting this power becomes negative, which was also discovered in previous research with another energy model. We also find that the neuron’s energy consumption during bursting is minimal. Especially in the spontaneous state without stimulation, the total energy con- sumption (2.152 9 10-7 J) during 30 s of bursting is very similar to the biological energy consumption (2.468 9 10-7 J) during the generation of a single action potential, as shown in Wang et al. (Neural Plast 2017, 2017a). Our results suggest that this property of low energy consumption could simply be the consequence of the biophysics of generating bursts, which is consistent with the principle of energy minimization. -

Characteristics of Hippocampal Primed Burst Potentiation in Vitro and in the Awake Rat
The Journal of Neuroscience, November 1988, 8(11): 4079-4088 Characteristics of Hippocampal Primed Burst Potentiation in vitro and in the Awake Rat D. M. Diamond, T. V. Dunwiddie, and G. M. Rose Medical Research Service, Veterans Administration Medical Center, and Department of Pharmacology, University of Colorado Health Sciences Center, Denver, Colorado 80220 A pattern of electrical stimulation based on 2 prominent (McNaughton, 1983; Lynch and Baudry, 1984; Farley and Al- physiological features of the hippocampus, complex spike kon, 1985; Byrne, 1987). Of these, a phenomenontermed long- discharge and theta rhythm, was used to induce lasting in- term potentiation (LTP) has received extensive attention be- creases in responses recorded in area CA1 of hippocampal causeit sharesa number of characteristicswith memory. For slices maintained in vitro and from the hippocampus of be- example, LTP has a rapid onset and long duration and is having rats. This effect, termed primed burst (PB) potentia- strengthenedby repetition (Barnes, 1979; Teyler and Discenna, tion, was elicited by as few as 3 stimuli delivered to the 1987). In addition, the decay of LTP is correlated with the time commissural/associational afferents to CAl. The patterns courseof behaviorally assessedforgetting (Barnes, 1979; Barnes of stimulus presentation consisted of a single priming pulse and McNaughton, 1985). The similarity of LTP and memory followed either 140 or 170 msec later by a high-frequency has fostered an extensive characterization of the conditions un- burst of 2-10 pulses; control stimulation composed of un- derlying the initiation of LTP, as well as its biochemical sub- primed high-frequency trains of up to 10 pulses had no en- strates (Lynch and Baudry, 1984; Wigstrijm and Gustafsson, during effect. -

The Logic Behind Neural Control of Breathing Pattern Alona Ben-Tal 1, Yunjiao Wang2 & Maria C
www.nature.com/scientificreports OPEN The logic behind neural control of breathing pattern Alona Ben-Tal 1, Yunjiao Wang2 & Maria C. A. Leite3 The respiratory rhythm generator is spectacular in its ability to support a wide range of activities and Received: 10 January 2019 adapt to changing environmental conditions, yet its operating mechanisms remain elusive. We show Accepted: 29 May 2019 how selective control of inspiration and expiration times can be achieved in a new representation of Published: xx xx xxxx the neural system (called a Boolean network). The new framework enables us to predict the behavior of neural networks based on properties of neurons, not their values. Hence, it reveals the logic behind the neural mechanisms that control the breathing pattern. Our network mimics many features seen in the respiratory network such as the transition from a 3-phase to 2-phase to 1-phase rhythm, providing novel insights and new testable predictions. Te mechanism for generating and controlling the breathing pattern by the respiratory neural circuit has been debated for some time1–6. In 1991, an area of the brainstem, the pre Bötzinger Complex (preBötC), was found essential for breathing7. An isolated single PreBötC neuron could generate tonic spiking (a non-interrupted sequence of action potentials), bursting (a repeating pattern that consists of a sequence of action potentials fol- lowed by a time interval with no action potentials) or silence (no action potentials)8. Tese signals are transmit- ted, through other populations of neurons, to spinal motor neurons that activate the respiratory muscle4,6. Te respiratory muscle contracts when it receives a sequence of action potentials from the motor neurons and relaxes when no action potentials arrive9. -
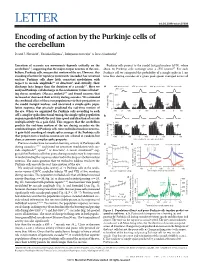
(2015) Encoding of Action by the Purkinje Cells of the Cerebellum
LETTER doi:10.1038/nature15693 Encoding of action by the Purkinje cells of the cerebellum David J. Herzfeld1, Yoshiko Kojima2, Robijanto Soetedjo2 & Reza Shadmehr1 Execution of accurate eye movements depends critically on the Purkinje cells project to the caudal fastigial nucleus (cFN), where cerebellum1–3, suggesting that the major output neurons of the cere- about 50 Purkinje cells converge onto a cFN neuron17. For each bellum, Purkinje cells, may predict motion of the eye. However, this Purkinje cell we computed the probability of a simple spike in 1-ms encoding of action for rapid eye movements (saccades) has remained time bins during saccades of a given peak speed, averaged across all unclear: Purkinje cells show little consistent modulation with respect to saccade amplitude4,5 or direction4, and critically, their discharge lasts longer than the duration of a saccade6,7.Herewe a 400° per s saccade 650° per s saccade 400° per s saccade 650° per s saccade analysed Purkinje-cell discharge in the oculomotor vermis of behav- 500° per s 8,9 200 ing rhesus monkeys (Macaca mulatta) and found neurons that N12 increased or decreased their activity during saccades. We estimated the combined effect of these two populations via their projections to 100 the caudal fastigial nucleus, and uncovered a simple-spike popu- lation response that precisely predicted the real-time motion of 0 the eye. When we organized the Purkinje cells according to each –1000 100 0 100 0 100 0 100 cell’s complex-spike directional tuning, the simple-spike population b 500° per s response predicted both the real-time speed and direction of saccade multiplicatively via a gain field. -

Analysis of Complex Bursting in Cortical Pyramidal Neuron Modelsଝ
Neurocomputing 32}33 (2000) 181}187 Analysis of complex bursting in cortical pyramidal neuron modelsଝ Adam Kepecs*, Xiao-Jing Wang Volen Center for Complex Systems, Brandeis University, Waltham, MA 02254, USA Accepted 13 January 2000 Abstract Burst "ring is a prominent feature of cortical pyramidal cells and is thought to have signi"cant functional roles in reliable signaling and synaptic plasticity. Modeling studies have successfully elucidated possible biophysical mechanisms underlying complex bursting in pyr- amidal cells. Based on these results (Pinsky, Rinzel, J. Comput. Neurosci. 1 (1994) 39}60), we have built a simpli"ed two-compartment burst model. Using the fast- and slow-variable analysis method, we show that complex bursting is an instance of square-wave bursting, where the dendritic slow potassium conductance is the single slow variable. The coupling parameters between the two compartments change the topological class of bursting thereby altering the "ring patterns of the neuron. These results explain the diverse set of "ring patterns seen with di!erent dendritic morphologies (Mainen, Sejnowski, Nature 382 (1996) 363}366). ( 2000 Elsevier Science B.V. All rights reserved. Keywords: Phase-plane analysis; Complex burst; Bifurcation diagram 1. Introduction Pyramidal cells in many cortical areas "re stereotyped bursts of action potentials termed complex spikes or complex bursts. These bursts consist of 2}7 action poten- tials occurring in a &30 ms window. First observed in hippocampal single-unit extracellular recordings, these were termed &complex spikes' and were later identi"ed by intracellular techniques. Burst "ring is thought to play an important role in reliable signaling [5,15] and synaptic plasticity [2]. -

Abnormal High-Frequency Burst Firing of Cerebellar Neurons in Rapid-Onset Dystonia-Parkinsonism
The Journal of Neuroscience, August 27, 2014 • 34(35):11723–11732 • 11723 Neurobiology of Disease Abnormal High-Frequency Burst Firing of Cerebellar Neurons in Rapid-Onset Dystonia-Parkinsonism Rachel Fremont,* D. Paola Calderon,* X Sara Maleki, and Kamran Khodakhah Dominick P. Purpura Department of Neuroscience, Albert Einstein College of Medicine, Bronx, New York 10461 Loss-of-function mutations in the ␣3 isoform of the Na ϩ/K ϩ ATPase (sodium pump) are responsible for rapid-onset dystonia parkin- sonism (DYT12). Recently, a pharmacological model of DYT12 was generated implicating both the cerebellum and basal ganglia in the disorder. Notably, partially blocking sodium pumps in the cerebellum was necessary and sufficient for induction of dystonia. Thus, a key question that remains is how partially blocking sodium pumps in the cerebellum induces dystonia. In vivo recordings from dystonic mice revealed abnormal high-frequency bursting activity in neurons of the deep cerebellar nuclei (DCN), which comprise the bulk of cerebellar output. In the same mice, Purkinje cells, which provide strong inhibitory drive to DCN cells, also fired in a similarly erratic manner. In vitro studies demonstrated that Purkinje cells are highly sensitive to sodium pump dysfunction that alters the intrinsic pacemaking of these neurons, resulting in erratic burst firing similar to that identified in vivo. This abnormal firing abates when sodium pump function is restored and dystonia caused by partial block of sodium pumps can be similarly alleviated. These findings suggest that persistent high-frequency burst firing of cerebellar neurons caused by sodium pump dysfunction underlies dystonia in this model of DYT12. Key words: basal ganglia; cerebellum Introduction sonism, otherwise referred to as DYT12 (de Carvalho et al., 2004; Dystonia is the third most common movement disorder after Brashear et al., 2007). -

Effects of Autapse and Channel Blockage on Firing Regularity in a Biological Neuronal Network
Rukiye UZUN and Mahmut OZER / IU-JEEE Vol. 17(1), (2017), 3069-3073 Effects of Autapse and Channel Blockage on Firing Regularity in a Biological Neuronal Network Rukiye UZUN1, Mahmut OZER 1 1Bulent Ecevit University, Zonguldak, Turkey [email protected], [email protected] Abstract: In this paper; the effects of autapse (a kind of self-synapse formed between the axon of the soma of a neuron and its own dendrites) and ion channel blockage on the firing regularity of a biological small-world neuronal network, consists of stochastic Hodgkin-Huxley neurons, are studied. In this study, it is assumed that all of the neurons on the network have a chemical autapse and a constant membrane area. Obtained results indicate that there are different effects of channel blockage and parameters of the autapse on the regularity of the network, thus on the temporal coherence of the network. It is found that the firing regularity of the network is decreased with the sodium channel blockage while increased with potassium channel blockage. Besides, it is determined that regularity of the network augments with the conductance of the autapse. Keywords: Autapse, channel blocking, ion channel noise, small-world neuronal netwoks. 1. Introduction investigated the effect of ion channel noise on dynamics of neuron in the presence of autapse based on a stochastic The nervous system, having a complex biologic Hodgkin-Huxley (HH) model. They found that autapse is structure, comprises of billions of nerve cells (neurons) reduced the spontaneous spiking activity of neuron at and synaptic connections between them [1]. In this characteristic frequencies by inducing bursting firing and complex structure, it is believed that the signal multimodal interspike interval distribution. -

Multiple Mechanisms of Bursting in a Conditional Bursting Neuron
The Journal of Neuroscience, July 1987, 7(7): 2113-2128 Multiple Mechanisms of Bursting in a Conditional Bursting Neuron Ronald M. Harris-Warrick and Robert E. Flamm” Section of Neurobiology and Behavior, Seeley G. Mudd Hall, Cornell University, Ithaca, New York 14853 The anterior burster (AB) neuron in the stomatogastric gan- many conductancesactive at appropriate times during the burst. glion of the spiny lobster, /Janu/ifus interruptus, is a condi- The current evidence suggeststhat the ionic mechanismsfor tional burster in the pyloric motor circuit. Bath application of bursting vary between different cells. the monoamines dopamine, serotonin, and octopamine in- Conditional bursters are a secondclass of endogenouslyburst- duces rhythmic bursting pacemaker potentials in a silent, ing neurons. In these cells, the ionic mechanismsthat generate synaptically isolated AB cell. However, each amine produces rhythmic activity must be activated by unpatterned modulatory a unique and characteristic burst shape, resulting from dif- input; when isolated, these cells lose the ability to burst. A ferent ionic dependences of the burst mechanisms. Bursting number of studieshave analyzed the ionic mechanismsof burst induced by serotonin or octopamine is critically dependent induction and maintenancein conditional bursting cells (Tazaki upon sodium entry through tetrodotoxin-sensitive channels; and Cooke, 1979, 1983; Gola and Selverston, 1981; Bensonand dopamine-induced bursting is not TTX-sensitive. Dopamine- Cooke, 1984; Berlind, 1985). Most studies of the induction of induced bursting is abolished when extracellular calcium is conditional bursting have focusedon a singlemajor conductance reduced to 25% of normal; serotonin- and octopamine-in- changein the affected cell (Barker and Smith, 1976; Bensonand duced bursts continue in this saline, although they are abol- Cooke, 1984; Nagy et al., 1984, 1985; McCormick and Prince, ished in salines with calcium reduced to 10% or less of 1986). -

Hippocampal Theta Bursting and Waveform Shape Reflect CA1 Spiking Patterns
bioRxiv preprint doi: https://doi.org/10.1101/452987; this version posted October 25, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Title Hippocampal theta bursting and waveform shape reflect CA1 spiking patterns Authors and Affiliations 1 1,2,3 Scott Cole , Bradley Voytek 1 2 3 Neurosciences Graduate Program, Department of Cognitive Science, Halıcıoğlu Data Science Institute, University of California, San Diego, La Jolla, CA, 92093, USA Acknowledgements We thank Thomas Donoghue, Richard Gao, Tammy Tran, and Roemer van der Meij for invaluable discussion. S.R.C. is supported by the National Science Foundation Graduate Research Fellowship Program and the University of California, San Diego Chancellor’s Research Excellence Scholarship. B.V. is supported by a Sloan Research Fellowship, the Whitehall Foundation (2017-12-73), and the National Science Foundation (1736028). Author contributions S.R.C. performed the analyses. S.R.C. and B.V. designed the research and wrote the paper. Competing interests The authors declare no competing interests. Abstract Brain rhythms are nearly always analyzed in the spectral domain in terms of their power, phase, and frequency. While this conventional approach has uncovered spike-field coupling, as well as correlations to normal behaviors and pathological states, emerging work has highlighted the physiological and behavioral importance of multiple novel oscillation features. Oscillatory bursts, for example, uniquely index a variety of cognitive states, and the nonsinusoidal shape of oscillations relate to physiological changes, including Parkinson's disease. -

Ghostbursting: the Effects of Dendrites on Spike Patterns
Neurocomputing 44–46 (2002) 127–132 www.elsevier.com/locate/neucom Ghostbursting: the e"ects of dendrites on spike patterns Carlo R. Lainga;∗, Brent Doirona;b , AndrÃe Longtina , Len Malerb aDepartment of Physics, University of Ottawa, 150 Louis Pasteur, Ottawa, Ont., Canada K1N 6N5 bDepartment of Cellular and Molecular Medicine, University of Ottawa, 451 Smyth Road, Ottawa, Ont., Canada K1H 8M5 Abstract We present a two-compartment model of a pyramidal cell from the electrosensory lateral line lobe of weakly electric ÿsh. These cells undergo a complex form of bursting, previously labeled “ghostbursting”, and our model reproduces many aspects of experimental recordings. We analyze the e"ects of varying both the conductance between the somatic and dendritic compartments in the model, and the ratio of somatic to whole-cell areas, and ÿnd that both must have moderate values for bursting to occur. This is explained in terms of the previously elucidated burst mechanism. c 2002 Published by Elsevier Science B.V. Keywords: Bursting; Pyramidal cell; Bifurcation; Compartmental model 1. Introduction Recently, a novel bursting mechanism was identiÿed in pyramidal cells in the elec- trosensory lateral line lobe (ELL) of the gymnotiform weakly electric ÿsh Apteronot- nus leptorhynchus [2,3,10,14]. Experimental in vitro recordings from these cells [10,14] show that under constant current injection they ÿre regular bursts of action potentials. One of the most striking aspects of these bursts is that the interspike intervals (ISIs) decrease monotonically during a burst, i.e. the instantaneous frequency (reciprocal of the current ISI) increases monotonically through the burst, and the burst ends with a high-frequency doublet. -

Chaotic Bursting in Single Neurons
Chaotic Bursting in Single Neurons Jon Newman¤ Department of Bioengineering Georgia Institute of Technology, Atlanta, GA 30332-0430, U.S.A (Dated: December 7, 2008) It has become fashionable to report on the existence of deterministic chaos in biophysical neuron models. Much of the time, the importance of this behavior is given as self-evident. Moreover, it is rarely transparent whether these studies actually investigate a chaotic behavior since a full mathematical analysis is not pursued. This report revisits a biophysical paper reporting chaotic bursting in an intrinsically bursting neuron model ref. [1]. Using cycle-expansion techniques, we determine whether these models in fact produce chaotic oscillations and how these oscillations come to be. We related these results to biological mechanisms that may be responsible for the chaotic dynamics in neurons. Finally, we investigate how likely chaotic bursting is to play a functional role in single neurons given the presence of large noise in the vast majority of biological systems. PACS numbers: 87.19.ll, 05.45.Ac, 05.10.{a Keywords: neuronal bursting, chaos, cycle-expansions 0. DISCLAIMER II. STATE SPACE ANALYSIS OF BURSTING Dynamical bursting models are a system of ordinary di®erential equations of the form This is a term report for the class `Chaos and What To Do About It' taught by Predrag Cvitanovi¶cat the Georgia x_ = f(x; y); (1) Institute of Technology during the Fall semester of 2008. y_ = ²g(x; y); x 2 Rm; y 2 Rn; (2) The content has not passed any rigorous peer review and may contain errors. where 0 · ² is a small parameter.