The Future of MS Treatments
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ectrims Meeting November 2015
SUPPLEMENT TO HIGHLIGHTS FROM THE ECTRIMS MEETING NOVEMBER 2015 Ocrelizumab May Reduce Disability Progression in People With Primary Progressive MS Data support the hypothesis that B cells are central to the underlying biology of MS. BARCELONA—In people with primary progressive Overall, the incidence of adverse events associated multiple sclerosis (MS), treatment with ocrelizumab may with ocrelizumab was similar to that of placebo. The most significantly reduce the progression of clinical disability common adverse events were mild-to-moderate infusion- sustained for at least 12 weeks, compared with placebo, related reactions. The incidence of serious adverse events according to results from a pivotal phase III study presented associated with ocrelizumab, including serious infections, at the 31st Congress of the European Committee for Treat- was also similar to that of placebo. ment and Research in Multiple Sclerosis (ECTRIMS). In the “People with the primary progressive form of MS typi- study, which is called ORATORIO, clinical disability was cally experience symptoms that continuously worsen after measured by the Expanded Disability Status Scale (EDSS). the onset of their disease, and there are no approved treat- Ocrelizumab is an investigational, humanized mono- ments for this debilitating condition,” said Sandra Horn- clonal antibody designed to selectively target CD20-posi- ing, MD, Chief Medical Officer and Head of Global Product tive B cells. CD20-positive B cells are a type of immune cell Development for Genentech, the developer -

Monoclonal Antibodies As Neurological Therapeutics
pharmaceuticals Review Monoclonal Antibodies as Neurological Therapeutics Panagiotis Gklinos 1 , Miranta Papadopoulou 2, Vid Stanulovic 3, Dimos D. Mitsikostas 4 and Dimitrios Papadopoulos 5,6,* 1 Department of Neurology, KAT General Hospital of Attica, 14561 Athens, Greece; [email protected] 2 Center for Clinical, Experimental Surgery & Translational Research, Biomedical Research Foundation of the Academy of Athens (BRFAA), 11527 Athens, Greece; [email protected] 3 Global Pharmacovigilance, R&D Sanofi, 91385 Chilly-Mazarin, France; vid.stanulovic@sanofi.com 4 1st Neurology Department, Aeginition Hospital, National and Kapodistrian University of Athens, 11521 Athens, Greece; [email protected] 5 Laboratory of Molecular Genetics, Hellenic Pasteur Institute, 129 Vasilissis Sophias Avenue, 11521 Athens, Greece 6 Salpetriere Neuropsychiatric Clinic, 149 Papandreou Street, Metamorphosi, 14452 Athens, Greece * Correspondence: [email protected] Abstract: Over the last 30 years the role of monoclonal antibodies in therapeutics has increased enormously, revolutionizing treatment in most medical specialties, including neurology. Monoclonal antibodies are key therapeutic agents for several neurological conditions with diverse pathophysio- logical mechanisms, including multiple sclerosis, migraines and neuromuscular disease. In addition, a great number of monoclonal antibodies against several targets are being investigated for many more neurological diseases, which reflects our advances in understanding the pathogenesis of these -
Q3 Tnt Submission Revised Sheet.Xlsx
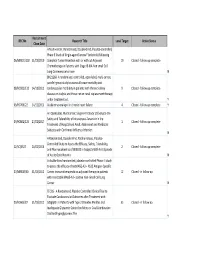
Recruitment REC No Research Title Local Target Active Status Close Date A Multi-center, Randomized, Double-blind, Placebo-controlled, Phase 3 Study of Single-agent Tarceva® (erlotinib) Following 06/MRE01/102 31/03/2010 Complete Tumor Resection with or without Adjuvant 10 Closed - follow up complete Chemotherapy in Patients with Stage IB-IIIA Non small Cell Lung Carcinoma who have N BH21260- A randomised, controlled, open-label, multi-centre, parallel-group study to assess all-cause mortality and 08/H0502/132 14/10/2011 cardiovascular morbidity in patients with chronic kidney 9 Closed - follow up complete disease on dialysis and those not on renal replacement therapy under treatment wit Y 09/H0706/22 14/12/2013 Aliskiren vs enalapril in chronic heart failure 4 Closed - follow up complete Y An OpenLabel, MultiCenter, Single Arm Study to Evaluate the Safety and Tolerability of Intravenous Zanamivir in the 09/H0802/125 27/01/2017 1 Closed - follow up complete Treatment of Hospitalised Adult, Adolescent and Paediatric Subjects with Confirmed Influenza Infection N A Randomized, Double-Blind, Parallel-Group, Placebo- Controlled Study to Assess the Efficacy, Safety, Tolerability, 12/SC/0527 26/02/2014 2 Closed - follow up complete and Pharmacokinetics of BIIB033 in Subjects With First Episode of Acute Optic Neuritis N A double-blind randomized, placebo-controlled Phase III study to assess the efficacy of recMAGE-A3 + AS15 Antigen -Specific 07/MRE08/40 31/05/2012 Cancer immunotherapeutic as adjuvant therapy in patients 12 Closed - in follow up -

WO 2016/025911 Al 18 February 2016 (18.02.2016)
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/025911 Al 18 February 2016 (18.02.2016) (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 9/14 (2006.01) A61K 9/16 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 38/00 (2006.01) A61K 9/19 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 39/00 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/US20 15/045402 KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (22) International Filing Date: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 14 August 2015 (14.08.2015) PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 62/037,492 14 August 2014 (14.08.2014) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicants: BROWN UNIVERSITY [US/US]; 47 TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, George Street, Providence, RI 02912 (US). -

Company Data, Credit Suisse Analysis Slide 4 September 2013 Our Top-10 Sound Bites/Observation/Questions/Focal Points for the Conference(Cont.)
Global Biotechnology and Pharmaceuticals 24th September 2013 Multiple Sclerosis – ECTRIMS 2013 Prep-Pack It’s A Revolution! But Just How Big? Research Analysts Ravi Mehrotra, Ph.D. 212-325-3487 [email protected] EU Pharma Team Koon Ching, Ph.D. Vamil Divan Lee Kalowski 44 20 7888 0304 212 325 6286 212 538 5394 212-325-3777 [email protected] [email protected] [email protected] [email protected] Ari Jahja Ronak Shah, Pharm.D., CFA Anuj Shah Jason Kantor 212 325 0767 212 325 9799 212 325 6931 415 249 7942 [email protected] [email protected] [email protected] [email protected] DISCLOSURE APPENDIX CONTAINS IMPORTANT DISCLOSURES, ANALYST CERTIFICATIONS, INFORMATION ON TRADE ALERTS, ANALYST MODEL PORTFOLIOS AND THE STATUS OF NON-U.S ANALYSTS. U.S. Disclosure: Credit Suisse does and seeks to do business with companies covered in its research reports. As a result, investors should be aware that the Firm may have a conflict of interest that could affect the objectivity of this report. Investors should consider this report as only a single factor in making their investment decision. Evolution AND Revolution! As a humorous point/observation, we find the titles of the two Biogen sponsored symposium to be held at ECTRIMS very interesting: 10/2 17.15 “Biogen Idec: Reinventing MS care – Evolution” and 10/3 07.45 “Biogen Idec: Reinventing MS care – Revolution”. Umm, we wonder where they got that inspiration from… Slide 2 September 2013 Table of Contents: It’s A Revolution! At ECTRIMS 2013, we are focused on these particular areas: 1) Dynamics of Tecfidera’s launch - Warehoused vs. -

09/H0402/92 20080763 Panitumumab Vs C
Recruitment Recruitment Rec Number Name of Trial Target End Date Trial Status Target met ? 20080763 Panitumumab vs Cetuximab in 09/H0402/92 Colorectal Cancer 10 05/01/2015 Closed - Follow Up Complete Y BO21004 - RO5072759 Safety and Efficacy 09-WSE04-55 Chemo Combo in untreated CLL 5 01/04/2022 Closed - In Follow Up Y Phase 3 Study-Tivozanib in Patients with 09/H0903/51 Advanced Renal Cell Carcinoma 5 05/01/2015 Closed - Follow Up Complete N LDK378-Dose Escalation/Expansion study in 10/MRA00/84 patients with ALK+ tumour 2 31/05/2016 Closed - In Follow Up Y Phase I Study of NY-ESO-1 +AS15 in 10/H0806/106 Melanoma Version 1 4 05/01/2015 Closed - Follow Up Complete N BH21260- Mircera? vs. reference ESAs in CKD 08/H0502/133 patients (SUB STUDY) 10 01/01/2016 Withdrawn N/A R-CHOP vs G-CHOP in previously untreated 11/EM/0225 patients with DLBCL 5 01/02/2018 Closed - In Follow Up N 11/NI/0147 SJM Portico 23-TF EU 12 30/11/2014 Closed - In Follow Up Y A UK Phase IIIB expanded access study of 11/SC/0499 INC424 in myelofibrosis 3 13/07/2015 Closed - Follow Up Complete Y 11/AL/0393 Synta 9090-08 20 04/12/2014 Closed - Follow Up Complete N 12/LO/0806 Aflibercept SQoP (AFLIBC06097) 5 31/12/2015 Closed - In Follow Up Y MASERATI 100 Permacol Collagen Paste 12-LO-0928 Study 10 30/04/2015 Closed - In Follow Up Y SafeHer: Safety study with subcutaneous 12/SC/0139 Trastuzumab in breast cancer 10 23/09/2016 Open N/A Safety and tolerability of ONO-4059 in 12/EM/0204 patients with NHL & CLL 7 15/07/2015 Closed - In Follow Up Y Clinical trial of an investigational -

MS ECHO: Highlights from the Consortium of Multiple Sclerosis Centers 2015 Annual Meeting
MS ECHO: Highlights from the Consortium of Multiple Sclerosis Centers 2015 Annual Meeting Gary Stobbe, MD Medical Director, MS Project ECHO Clinical Assistant Professor, UW Neurology Conflicts of Interest • Dr. Stobbe has no conflicts of interest to disclose Objectives • Review topics of interest presented in posters and oral presentations at the CMSC 2015 Annual Meeting as well as AAN 2015 Annual Meeting • Discuss the importance of presented research as related to current clinical practice Benefits of Alemtuzumab when switching DMTs • Prospective, open label study (60 patients) – Median EDSS 5-5.5 – Short-term follow-up group (median – 10 mo; n-30) and long- term group (median – 82 mo; n-30) • EDSS improved by 0.4 in short-term group and by 1.0 in long-term group – Improvement seen with all prior DMTs (interferon-beta & glatiramer – 0.9, 82 mo; fingolimod – 0.4, 12 mo; natalizumab – 0.5, 49 mo) • Limitations – no MRI data; no patient-reported outcomes Hunter SF et al. Daclizumab HYP vs interferon Beta-1a in RRMS: primary results of the DECIDE study • Daclizumab high yield process (DAC HYP) – humanized monoclonal antibody against CD25, results in reversible modulation of IL-2 signaling • Study design – randomized, double-blind, active- controlled DAC HYP 150 mcg sq q4wks vs IFNβ-1a 30 mcg IM qweekly for 96-144 wks; 1° endpoint – annualized relapse rate (ARR) • Demographics – N=1841; mean age 36.3; mean EDSS 2.5; 41% prior DMT use Kaufman M. et al. Daclizumab HYP vs interferon Beta-1a in RRMS: primary results of the DECIDE study (cont.) • Results – – 45% reduction in the ARR (p<0.0001) – 41% reduction in proportion of patients that relapsed (p<0.0001) – 54% reduction in # of new/enlarging T2 lesions (p<0.0001) – 3-month confirmed disability reduction of 16% (p=0.158) – Improvement seen in all components of the MSFC and the SDMT (P=0.0274) Kaufman M. -

WO 2014/144600 A2 18 September 2014 (18.09.2014) P O P CT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2014/144600 A2 18 September 2014 (18.09.2014) P O P CT (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 39/395 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/US20 14/029077 KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, (22) International Filing Date: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 14 March 2014 (14.03.2014) OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (25) Filing Language: English TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, (26) Publication Language: English ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 61/802,343 15 March 2013 (15.03.2013) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, (72) Inventors; and UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, (71) Applicants : ROSCHKE, Viktor [US/US]; 4702 West TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, Virginia Avenue, Bethesda, MD 20814 (US). -

PDF of Antibody News
Antibody News You Should Know From: The Antibody Society <[email protected]> Sent: Monday, May 6, 2019 3:00 AM To: [email protected] Subject: Antibody News You Should Know View this email in your browser Antibody News You Should Know April 15 - May 1, 2019 Antibody-based therapeutics are entering clinical study at a rate of ~120 per year, and being approved in record numbers. Recent news about relevant events are summarized below. Follow the links to find more details. New (or close!) to the clinic: Antibody-based therapeutics that recently entered clinical studies or will soon Innovent Biologics, Inc., a world-class biopharmaceutical company that develops and commercializes high quality medicines, announced that the first patient in China has been successfully dosed in a Phase I clinical trial of IBI318. IBI318 is a recombinant fully human bispecific antibody targeting programmed cell death receptor-1 (PD-1) and programmed cell death ligand-1 (PD-L1). Innovent Biologics, is developing IBI318 in collaboration with Eli Lilly and Company. Innovent Biologics, Inc. also announced that the first patient has been successfully dosed in a Phase I clinical trial of IBI302 as a treatment of wet age-related macular degeneration (wet AMD). Antibody News You Should Know IBI302 is a novel recombinant fully human bispecific fusion protein targeting both vascular endothelium growth factor (VEGF) and complement proteins GEMoaB Monoclonals GmbH has initiated a multicenter, open-label, dose- escalating, Phase I trial with GEM3PSCA in patients with progressive disease after standard systemic therapy in cancers with positive PSCA marker. GEM3PSCA is a prostate stem cell antigen-targeted bispecific antibody engaging T-cells Harpoon Therapeutics, Inc., a clinical-stage immunotherapy company developing a novel class of T cell engagers, announced today that the first patient has been dosed with HPN536 in a Phase 1/2a clinical trial initially focused on ovarian cancer. -
ERANSKINA ANEXO I+G Proiektuen, Azterlan Klinikoen Eta Argitalpen Zientifikoen Zerrenda Proyectos De I+D, Estudios Clínicos Y Lista De Publicaciones Científicas 1
2018ko MEMORIA 2018 I+G+B jarduera Euskal Osasun Sistema Publikoan Actividad de I+D+i del Sistema Sanitario Público Vasco ERANSKINA ANEXO I+G proiektuen, azterlan klinikoen eta argitalpen zientifikoen zerrenda Proyectos de I+D, estudios clínicos y lista de publicaciones científicas 1. 2018an jarduneko proiektu lehiakorrak Proyectos competitivos activos en 2018 4 Índice de contenido 2. 2018an jarduneko proiektu lehiakorrak Eduki- Estudios clínicos activos en 2018 52 aurkibidea 3. Euskal osasun sistema publikoaren 2018ko argitalpenak Publicaciones 2018 del sistema sanitario público vasco 172 MEMORIA 2018 - ERANSKINA/ANEXO / 3 2018. urteari dagokion inbentario Se incluye en el presente inventario honetan, honako hauek ageri dira: relativo a 2018 las siguientes: • 2018ko jarduneko ikerketa-proiektuak, lehia-erregimeneko • Los proyectos de investigación activos durante el 2018, deialdien bidez eskuratuak eta Euskal Osasun Sistema obtenidos a través de convocatorias competitivas y que se Publikoan gauzatzen direnak. ejecutan en el Sistema Sanitario Público Vasco. Ordenado por centro y anualidad. • 2018ko jarduneko azterlan klinikoak; barne hartzen ditu medikamentuen eta produktu sanitarioen azterlan klinikoak, • Los estudios clínicos activos en 2018, incluye ensayos baimendu osteko azterlanak eta behaketa-motako clínicos de medicamentos y productos sanitarios, estudios azterlanak. Azterlan horietako asko hainbat zentrotan post-autorización y estudios observacionales. Muchos de gauzatzen dituzte aldi berean, Osakidetzaren zerbitzuetako estos estudios son multicéntricos y se ejecutan en varias de zenbait erakundetan. las organizaciones de servicios de Osakidetza. Ordenado por código identificativo y anualidad. • 2018ko argitalpen zientifikoak, hala nola honako hauek: abstract of Item Published, article, correction, editorial • Publicaciones científicas de 2018, entre las que se incluyen material, letter, meeting abstract eta review. -

1 Freedman Review of Available Therapies
Review of Available Therapies Mark S. Freedman MSc MD FANA FAAN FRCPC Professor of Medicine (Neurology) University of Ottawa Sr. Scientist The Ottawa Hospital Research Institute Disclosures Receipt of research or educational grants: Genzyme Receipt of honoraria or consultation fees: Actelion, BayerHealthcare, BiogenIdec, Chugai, EMD Canada, Genzyme, Hoffman La-Roche, Novartis, Sanofi-Aventis, Teva Canada Innovation Member of a company advisory board, board of directors or other similar group: Actelion, BiogenIdec, Genzyme, Merck Serono, Novartis, Sanofi-Aventis, Teva Canada Innovation Participation in a company sponsored speaker’s bureau: Genzyme Existing & Emerging MS therapies Other Phase I Lymphocyte CS-0777 BIIB033 trafficking Phase II Idebenone Phase III Firategrast Interferons ONO-4641 ELND-002 Marketed Siponimod Ponesimod AZD5904 Peg IFNβ 1a IFNβ-1b Fingolimod GRC4039 IFNβ-1a im Natlizumab IFNβ-1a sc CCX-140 Azathioprine Laquinimod Novantrone AIN457 Immune BG12 regulation Teriflunomide GA Daclizumab Antiproliferative Secukinumab agents Cladribine THC:CBD NI-0801 GA generics X2 4-AP PiXantrone GA 40 tiw AlemtuzumabIPX-056 RPI-78M Ocrelizumab ATX-MS-1467 Ofatumumab Nerispirdine PI2301 LY-2127399 Vaccine, RTL1000 Symptomatic Tx tolerization Belimumab = Oral administration Cytolytic mAbs = Injectable mAb, monoclonal antibody; Tx, treatment Evolving Treatment Landscape in MS RRMS Progressive MS (SPMS/PPMS) Generic/Biosimilar RRMS mAb Cladribine Firategrast Fingolimod MD1003 Orals Teriflunomide NeXt Gen Fingolimod Laquinimod S1pRA Dimethyl NeXt Gen Fumarate Masitinib Siponimod Fumarates Approved 2013 2014 2015 2016 2017 2018+ GA GA 3TW Tcelna Injectables Peg IFNβ1a (non Mab) IFNβ1a IFNβ1b Biosimilar Generic GA? Interferon-β MitoXantrone Natalizumab Alemtuzumab Daclizumab Ocrelizumab Ofatumumab mAbs Natalizumab Secukinumab Anti-LINGO1 Remyelination rHIgM22 GSK239512 MS: Pathological vs. -

2017 MS Research Update Is Made Possible Through Contributions in Honor Of: Dr
MAY 2017 RESEARCH MS UPDATE Written and compiled by Michelle Fabian, MD and Stephen Krieger, MD Includes additional material by MS Margaret M. McCormick, RN, BSN, MSCN RESEARCH Reviewed by Jack Burks, MD UPDATE Edited by Susan Courtney MSAA’s 2017 edition of the MS Research Update provides important new data on approved and experimental treatments for MS, and is a valuable resource to the entire MS community. Please note that this update gives an overview of the research behind the approved and experimental medications and therapies for the long-term treatment of multiple sclerosis. It does not include information on any symptom-management medications or therapies. For additional information about MS, symptoms and symptom management, as well as MSAA’s programs and services, please visit mymsaa.org or call (800) 532-7667. Questions to MSAA’s Client Services department may also be emailed to [email protected]. Additionally, please note that due to the timing of the national and international MS conferences, study data from the 2017 conferences could not be included in this writing. Information in this publication includes data presented at the 2016 conferences, as well as any important updates that occurred in early 2017. Please visit MSAA’s website at mymsaa.org for future summaries of 2017 conference highlights. The 2017 MS Research Update is made possible through contributions in honor of: Dr. Jules Kernan and Ms. Hannah Dennehy Lee and an anonymous supporter Multiple Sclerosis Association of America National Headquarters website: