Crystal Growth & Design 2007 Vol. 7, No. 12 2460–2466
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

On the Nature and Significance of Rarity in Mineralogy
1 1 REVISION #2—American Mineralogist—January 12, 2016 2 3 On the nature and significance of rarity in mineralogy 4 5 Robert M. Hazen1* and Jesse H. Ausubel2 6 1Geophysical Laboratory, Carnegie Institution, 5251 Broad Branch Road NW, Washington, D. C. 20015, USA. 7 2Program for the Human Environment, Rockefeller University, 1230 York Ave., New York, New York 10021, USA. 8 9 ABSTRACT 10 More than half of the >5000 approved mineral species are known from 5 or fewer localities 11 and thus are rare. Mineralogical rarity arises from different circumstances, but all rare mineral 12 species conform to one or more of 4 criteria: (1) P-T-X range: minerals that form only under 13 highly restricted conditions in pressure-temperature-composition space; (2) Planetary constraints: 14 minerals that incorporate essential elements that are rare or that form at extreme conditions that 15 seldom occur in Earth’s near-surface environment; (3) Ephemeral phases: minerals that rapidly 16 break down under ambient conditions; and (4) Collection biases: phases that are difficult to 17 recognize because they lack crystal faces or are microscopic, or minerals that arise in lithological 18 contexts that are difficult to access. Minerals that conform to criterion (1), (2), or (3) are 19 inherently rare, whereas those matching criterion (4) may be much more common than 20 represented by reported occurences. 21 Rare minerals, though playing minimal roles in Earth’s bulk properties and dynamics, are 22 nevertheless of significance for varied reasons. Uncommon minerals are key to understanding 23 the diversity and disparity of Earth’s mineralogical environments, for example in the prediction 24 of as yet undescribed minerals. -

Ternary Solubility Diagrams at 270&Nbsp
Fluid Phase Equilibria 437 (2017) 1e13 Contents lists available at ScienceDirect Fluid Phase Equilibria journal homepage: www.elsevier.com/locate/fluid 2þ 2þ Partitioning of Co and Mn into meridianiite (MgSO4$11H 2O): Ternary solubility diagrams at 270 K; cation site distribution determined by single-crystal time-of-flight neutron diffraction and density functional theory * A.D. Fortes a, b, c, , I.G. Wood b, K.A. Hudson-Edwards c, M.J. Gutmann a a ISIS Facility, STFC Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Chilton, Didcot, Oxfordshire, OX11 0QX, UK b Department of Earth Sciences, University College London, Gower Street, London WC1E 6BT, UK c Department of Earth and Planetary Sciences, Birkbeck, University of London, Malet Street, London WC1E 7HX, UK article info abstract 2þ Article history: We have grown single crystals of M SO4 hydrates at 270 K from aqueous solutions in the ternary Received 14 October 2016 systems CoSO4eMgSO4eH2O and MnSO4eMgSO4eH2O. These systems exhibit broad stability fields for a Received in revised form triclinic undecahydrate on the Mg-rich side (i.e., Co- or Mn-bearing meridianiite solid solutions) and 2 January 2017 stability fields for monoclinic heptahydrates on the Mg-poor side (i.e., Mg-bearing solid solutions of Accepted 8 January 2017 bieberite or mallardite). The solubility curves and distribution coefficients, describing the partitioning of Available online 9 January 2017 þ M2 ions between liquid and solid phases, have been determined by thermo-gravimetric and spectro- þ scopic techniques. A subset of M2 SO $11H O specimens were selected for single-crystal time-of-flight Keywords: 4 2 Meridianiite neutron diffraction analysis in order to evaluate preferential occupancy of symmetry-inequivalent co- fi Epsomite ordination polyhedra in the structure. -

Phase Transition Pathways of the Hydrates of Magnesium Sulfate in the Temperature Range 50°Cto5°C: Implication for Sulfates on Mars Alian Wang,1 John J
JOURNAL OF GEOPHYSICAL RESEARCH, VOL. 114, E04010, doi:10.1029/2008JE003266, 2009 Click Here for Full Article Phase transition pathways of the hydrates of magnesium sulfate in the temperature range 50°Cto5°C: Implication for sulfates on Mars Alian Wang,1 John J. Freeman,1 and Bradley L. Jolliff1 Received 19 September 2008; revised 5 December 2008; accepted 5 January 2009; published 25 April 2009. [1] Dehydration/rehydration experiments were conducted on pure Mg-sulfates and mixtures of epsomite with Ca-sulfates, Fe-sulfates, Fe-oxide, and Fe-hydroxide. The goal was to investigate the stabilities and phase transition pathways of Mg-sulfate hydrates, under temperature and relative humidity conditions relevant to Mars, as a function of starting structure and coexisting species. Two pathways were found to form Mg-sulfate monohydrates between 5°C and 50°C through dehydration of epsomite or hexahydrite. Two polymorphs of Mg-sulfate monohydrates were characterized in this study. It is important to distinguish among these phases on Mars because they have different formation conditions that have the potential to provide additional information on surface and subsurface geologic processes. We found that Mg-sulfates with moderate hydration states (especially starkeyite and amorphous Mg-sulfates) can be very stable under current Martian surface conditions. On the basis of NIR spectral features, these phases are good candidates for polyhydrated sulfates identified on Mars by OMEGA and CRISM; thus, they may contribute to the high hydrogen concentrations found by the neutron spectrometer on the orbiting Odyssey spacecraft. Our experiments indicate that the maximum number of water molecules per SO4 held by the amorphous Mg-sulfate structure is three. -

Chapter5 Crystallization and Characterization of a New
Propositions accompanying the thesis of F. Elif Genceli Scaling-Up Eutectic Freeze Crystallization 1. In heat flow calculations of crystallization on a cold surface [Pronk, P. et al.; Chemical Engineering Science, 2006, 61, p.4354-4362; Mershmann, A.; Crystallization Technology Handbook, 2001], it is frequently assumed that all heat of crystallization is transferred to the cold side which is not necessarily true (Chapter 7). 2. The Raman spectra presented in the work of Freeman et al. [Freeman J.J.; Wang, A.; Jolliff B.L.; 38th Lunar and Planetary Science Conference, 2007, No. 1338, p.1197] are not those of MgSO4.11H2O (Meridianiite) as they claim, but are spectra of crystal samples which have already been dehydrated into a magnesium sulfate with a lower hydrate content (Chapter 5-6). 3. It is surprising that no care was ever given since 1837 [Fritzsche, C.J.; Poggendorff’s Annalen, 1837, 42, 577-580] to determine the exact phase diagram of MgSO4 aqueous solution nor to determining the crystal structure of the magnesium sulfate phase formed at lower temperatures, despite the fact that the solution has been used in many crystallization applications as the model solution. [e.g.: Hogenboom, D.L.; Kargel, J.S.; Ganasan, J.P.; Lee, L.; Icarus. 1995, 115, 258-277] (Chapter 5). 4. It is easier to make nano-crystals from supercritical CO2 [Jung, J.; Perrut, M.; J. Supercrit. Fluids, 2001, 20, 3, 179-219] than by arrested precipitation from solution. 5. Putting CO2 underground is not a sustainable solution to our energy problem. 6. It is surprising, even paradoxical, that results in highly prestigious journals are not necessarily more reliable than those in second-tier journals. -

Meridianiite Detected in Ice
Journal of Glaciology, Vol. 55, No. 189, 2009 117 Meridianiite detected in ice F. Elif GENCELI,1 Shinichirou HORIKAWA,2 Yoshinori IIZUKA,2 Toshimitsu SAKURAI,2 Takeo HONDOH,2 Toshiyuki KAWAMURA,2 Geert-Jan WITKAMP1 1Process Equipment Section, Delft University of Technology, Leeghwaterstraat 44, 2628 CA Delft, The Netherlands E-mail: [email protected] 2Institute of Low Temperature Science, Hokkaido University, Sapporo 060-0819, Japan ABSTRACT. Inclusions affect the behavior of ice, and their characteristics help us understand the formation history of the ice. Recently, a low-temperature magnesium sulfate salt was discovered. This Á paper describes this naturally occurring MgSO4 11H2O mineral, meridianiite, derived from salt inclusions in sea ice of Lake Saroma, Japan and in Antarctic continental core ice. Its occurrence is Á confirmed by using micro-Raman spectroscopy to compare Raman spectra of synthetic MgSO4 11H2O with those of the inclusions. INTRODUCTION angle between the x and y crystallographic axes, ¼ 8 Seasonal sea ice strongly modulates global climate and eco- 62.598(1) ; logy through its effect on ocean albedo, ocean–atmosphere unit cell volume, V ¼ 699.54(3) A˚ 3; heat transfer and transfer of gases and particles (including nutrients) between the atmosphere and ocean. Small changes number of formula units in the crystallographic unit cell, ¼ in the atmosphere/ocean/ice composition ratio may lead to Z 2; significant changes in the nature of the sea-ice cover and its ¼ –3 calculated density, Dcalc 1.512 g cm ; and ecology. Investigations of mineral inclusions in sea ice are ¼ –1 expected to provide insights which may help in climate absorption coefficient, 0.343 mm (Genceli and studies. -

The Stability of Sulfate and Hydrated Sulfate Minerals Near Ambient Conditions and Their Significance in Environmental and Plane
Journal of Asian Earth Sciences 62 (2013) 734–758 Contents lists available at SciVerse ScienceDirect Journal of Asian Earth Sciences journal homepage: www.elsevier.com/locate/jseaes Review The stability of sulfate and hydrated sulfate minerals near ambient conditions and their significance in environmental and planetary sciences ⇑ I-Ming Chou a, , Robert R. Seal II a, Alian Wang b a U.S. Geological Survey, 954 National Center, Reston, VA 20192, USA b Department of Earth and Planetary Sciences and McDonnell Center for Space Sciences, Washington University, St. Louis, MO 63130, USA article info abstract Article history: Sulfate and hydrated sulfate minerals are abundant and ubiquitous on the surface of the Earth and also on Received 7 February 2012 other planets and their satellites. The humidity-buffer technique has been applied to study the stability of Received in revised form 5 November 2012 some of these minerals at 0.1 MPa in terms of temperature-relative humidity space on the basis of hydra- Accepted 12 November 2012 tion–dehydration reversal experiments. Updated phase relations in the binary system MgSO –H O are Available online 28 November 2012 4 2 presented, as an example, to show how reliable thermodynamic data for these minerals could be obtained based on these experimental results and thermodynamic principles. This approach has been Keywords: applied to sulfate and hydrated sulfate minerals of other metals, including Fe (both ferrous and ferric), Metal sulfate Zn, Ni, Co, Cd, and Cu. Hydrated sulfate minerals Humidity and temperature Metal–sulfate salts play important roles in the cycling of metals and sulfate in terrestrial systems, and Thermodynamics and kinetics the number of phases extends well beyond the simple sulfate salts that have thus far been investigated Terrestrial occurrence experimentally. -

Advances and Gaps in the Knowledge of Thermodynamics and Crystallography of Acid Mine Drainage Sulfate Minerals
MINERALOGY CHIMIA 2010, 64, No. 10 699 doi:10.2533/chimia.2010.699 Chimia 64 (2010) 699–704 © Schweizerische Chemische Gesellschaft Advances and Gaps in the Knowledge of Thermodynamics and Crystallography of Acid Mine Drainage Sulfate Minerals Juraj Majzlan* Abstract: Acidic and metal-rich waters produced by sulfide decomposition at mining sites are termed acid mine drainage (AMD). They precipitate a number of minerals, very often sulfates. The recent advances in thermodyna- mic properties and crystallography of these sulfates are reviewed here. There is a reasonable amount of data for the divalent (Mg, Ni, Co, Fe2+, Cu, Zn) sulfates and these data may be combined with and optimized by tempe- rature-relative humidity brackets available in the literature. For the sulfates with Fe3+, most data exist for jarosite; for other minerals and phases in this system, a few calorimetric studies were reported. No data whatsoever are available for the Fe2+-Fe3+ sulfates. A significant advance is the development of the Pitzer model for Fe3+-sulfate solutions and its confrontation with the available thermodynamic and solubility data. In summary, our knowledge about the thermodynamic properties of the AMD sulfates is unsatisfactory and fragmented. Keywords: Acid mine drainage Introduction population matches the quantities of the relatively recently.[12,13] A sizeable body of overall natural mass flow.[4] The mining of data accumulated during the last ten years, The ever-increasing demand for metals ores and salts does not dominate the an- partially because of interest in AMD, par- dictates the steady increase in the exploi- thropogenic mass flow but cannot be ne- tially because sulfate minerals have been tation of ore deposits, and, to a smaller glected. -
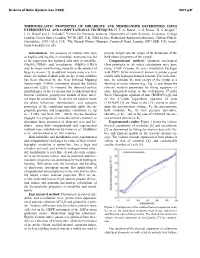
Thermoelastic Properties of Mirabilite and Meridianiite Determined Using Experimental and Computational Techniques
Science of Solar System Ices (2008) 9021.pdf THERMOELASTIC PROPERTIES OF MIRABILITE AND MERIDIANIITE DETERMINED USING EXPERIMENTAL AND COMPUTATIONAL TECHNIQUES. H. E. A. Brand1, A. D. Fortes1, K. S. Knight2, 3 I. G. Wood1 and L. Vočadlo1. 1Centre for Planetary Sciences, Department of Earth Sciences, University College London, Gower Street. London, WC1E 6BT, U.K. 2ISIS facility, Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire, OX11 0LA, U.K. 3The Natural History Museum, Cromwell Road, London, SW7 5BD, U.K (email; [email protected]). Introduction: The presence of soluble salts such provide insight into the origin of the behaviour of the as MgSO4 and Na2SO4 in chondritic meteorites has led bulk elastic properties of the crystal. to the suggestion that hydrated salts such as mirabilite Computational method: Quantum mechanical (Na2SO4·10H2O) and meridianiite (MgSO4·11H2O) (first principles or ab initio) calculations were done may be major rock-forming minerals in the mantles of using VASP (Vienna Ab initio Simulation Package) large icy moons [1], in addition to pure water ice. Evi- with PW91 GGA functional, known to produce good dence for hydrated alkali salts on the jovian satellites results with hydrogen-bonded systems. For each struc- has been observed by the Near Infrared Mapping ture, we calculate the total energy of the crystal as a Spectrometer (NIMS) instrument aboard the Galileo function of molar volume (e.g., Fig. 2) and obtain the space-craft [2][3]. To interpret the observed surface relevant material parameters by fitting equations of morphologies of the icy moons and to understand their state. Integrated forms of the well-known 3rd-order thermal evolution, geophysical models of their interi- Birch-Murnaghan equation of state (BMEOS3) [4] and ors must be constructed. -

Brine Evolution and Transport-Driven Fractionation of Ocean Fluids Within Europa’S Icy Shell
52nd Lunar and Planetary Science Conference 2021 (LPI Contrib. No. 2548) 1929.pdf BRINE EVOLUTION AND TRANSPORT-DRIVEN FRACTIONATION OF OCEAN FLUIDS WITHIN EUROPA’S ICY SHELL. M. G. Fox-Powell1, J. Semprich1, N. Ramkissoon1, J. J. Buffo2, S. D. Vance3, S. P. 1 1 1 Schwenzer , V. K. Pearson . AstrobiologyOU, Open University, Walton Hall, Milton Keynes, UK MK7 6AA, [email protected]; 2Dartmouth College, Hanover, NH, USA; 3Jet Propulsion Laboratory, California Institute of Technology, Pasadena, CA, USA. Introduction: Constraining the composition of magnesium were systematically varied independently Europa’s subsurface ocean is a vital step in between 0.001 and 0.499. The relative abundances of all understanding the moon’s evolution and potential other ions were kept constant, fixed according to the habitability. Models of water-rock interaction at model of [1]. The total salinities of the hypothetical Europa’s seafloor indicate that a sulfate-rich ocean oceans were also kept constant (here reported at should be expected [1,2], but recent Earth-based terrestrial seawater salinity; 34 g kg-1), to allow direct telescope observations suggest that sodium and comparison of solid phase formation temperatures as a magnesium-chloride salts, not sulfates, dominate function of fluid composition alone. geologically young regions where exchange with the subsurface may have taken place [3,4]. Fractional Results & Discussion: Equilibrium freezing. If crystallization of ocean fluids during freezing can act to ocean-derived fluids are transported in a manner that enrich chlorides within ice, even from sulfate- allows residual brines to remain in equilibrium with dominated fluids [5], offering the possibility to precipitated phases, MgCl2 minerals only form from 2+ 2- reconcile these views. -

Subsurface Water Oceans on Icy Satellites: Chemical Composition and Exchange Processes
Space Sci Rev (2010) 153: 485–510 DOI 10.1007/s11214-010-9646-y Subsurface Water Oceans on Icy Satellites: Chemical Composition and Exchange Processes Frank Sohl · Mathieu Choukroun · Jeffrey Kargel · Jun Kimura · Robert Pappalardo · Steve Vance · Mikhail Zolotov Received: 24 August 2009 / Accepted: 1 March 2010 / Published online: 13 April 2010 © Springer Science+Business Media B.V. 2010 Abstract The state of knowledge about the structure and composition of icy satellite interi- ors has been significantly extended by combining direct measurements from spacecraft, lab- oratory experiments, and theoretical modeling. The existence of potentially habitable liquid water reservoirs on icy satellites is dependent on the radiogenic heating of the rock compo- nent, additional contributions such as the dissipation of tidal energy, the efficiency of heat transfer to the surface, and the presence of substances that deplete the freezing point of liq- uid water. This review summarizes the chemical evolution of subsurface liquid water oceans, taking into account a number of chemical processes occuring in aqueous environments and F. Sohl () Institute of Planetary Research, German Aerospace Center (DLR), Rutherfordstr. 2, 12489 Berlin, Germany e-mail: [email protected] M. Choukroun · R. Pappalardo · S. Vance Jet Propulsion Laboratory, Caltech, MS 79-24, 4800 Oak Grove Drive, Pasadena, CA 91106, USA M. Choukroun e-mail: [email protected] R. Pappalardo e-mail: [email protected] S. Vance e-mail: [email protected] J. Kargel Department of Hydrology, University of Arizona, Tuscon, AZ 85721, USA e-mail: [email protected] J. Kimura Department of Cosmosciences, Hokkaido University, Nishi 8, Kita 10, Kita-Ku, Sapporo 060-0810, Japan e-mail: [email protected] M. -

An Experiment Recreates the Crust of the Moon Europa 14 March 2014
An experiment recreates the crust of the moon Europa 14 March 2014 the space missions observations suggest. Data also suggest that red materials are hydrated salts, mainly of magnesium sulfate (MgSO4). Volatile compounds like carbon dioxide (CO2), sulphur dioxide (SO2) and hydrogen peroxide (H2O2) have been also detected. Now, with all these premises, researchers at Centro de Astrobiología (CAB, INTA-CSIC) have developed an experiment to explain how these fluids evolve in their way between the deep reservoirs and the moon's surface. "Just like Earth's magma emerges to the surface, a similar phenomenon could occur in Europa. Although, in this case it would be a watery The scientists have simulated in the laboratory the cryomagma that would evolve and emerge extreme conditions of the fluid reservoirs in the crust of outwards from the interior of the icy moon," says Europa. Credit: CAB (INTA-CSIC) Victoria Muñoz Iglesias, one of this work's authors, published in the journal Geochimica et Cosmochimica Acta. Water, salts and gases dissolved in the huge ocean that scientists believe could exist below Europa´s icy crust can rise to the surface generating the enigmatic geological formations associated to red-tinged materials that can be seen on this Jupiter's satellite. This is confirmed by the experiment carried out in the laboratory with water, carbon dioxide and magnesium sulfate by researchers at Centro de Astrobiología (CAB). Scientists suspect that inside Europa, one of the icy moons of Jupiter, reservoirs of liquid water exists, the essential element for life on Earth. This theory emerged from information obtained on the Voyager and Galileo missions, which also registered fractures and `chaotic´ terrains associated to reddish materials, which contrast with the glacial white of the dominant water ice of Lenticulae terrain on the surface of Europa. -

74205834.Pdf
2+ 2+ Partitioning of Co and Mn into meridianiite (MgSO4·11H2O): ternary solubility diagrams at 270 K; cation site distribution determined by single- crystal time-of-flight neutron diffraction and density functional theory. ! A. D. Fortes,1,2,3,† I. G. Wood,2 K. A. Hudson-Edwards,3 and M. J. Gutmann1 ! 1ISIS Facility, STFC Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Chilton, Didcot, Oxfordshire, OX11 0QX, U.K. 2Department of Earth Sciences, University College London, Gower Street, London WC1E 6BT, U.K. 3Department of Earth and Planetary Sciences, Birkbeck, University of London, Malet Street, London WC1E 7HX, U.K. ! †Current permanent address, ISIS Facility, STFC Rutherford Appleton Laboratory. Corresponding author email [email protected] ! Abstract ! 2+ We have grown single crystals of M SO4 hydrates at 270 K from aqueous solutions in the ternary systems CoSO4–MgSO4–H2O and MnSO4–MgSO4–H2O. These systems exhibit broad stability fields for a triclinic undecahydrate on the Mg-rich side (i.e., Co- or Mn- bearing meridianiite solid solutions) and stability fields for monoclinic heptahydrates on the Mg-poor side (i.e., Mg-bearing solid solutions of bieberite or mallardite). The solubility curves and distribution coefficients, describing the partitioning of M2+ ions between liquid and solid phases, have been determined by thermo-gravimetric and spectroscopic techniques. 2+ A subset of M SO4·11H2O specimens were selected for single-crystal time-of-flight neutron diffraction analysis in order to evaluate preferential occupancy of symmetry-inequivalent coordination polyhedra in the structure. Considering the nearly identical dimensions of the first coordination shells, there is a surprising difference in the distribution of Co and Mn over the two available sites.