Super-Resolution Ribosome Profiling Reveals Unannotated Translation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Selfing Syndrome Overshadows Other Differences When Comparing
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.26.398016; this version posted November 27, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The selfing syndrome overshadows other differences when 2 comparing fitness across Capsella species 3 4 5 Marion Orsucci1, Theofilos Vanikiotis2, Maria Guerrina1, Tianlin Duan1, Sylvain Glémin3, Martin 6 Lascoux1 7 8 1 Department of Ecology and Genetics, Evolutionary Biology Centre and Science for Life 9 Laboratory, Uppsala University, 75236 Uppsala, Sweden 10 2 Department of Biological Applications & Technology, University of Ioannina, Leof. S. 11 Niarchou GR-451 10, Ioannina, Greece 12 3 UMR CNRS 6553 ECOBIO, Campus Beaulieu, bât 14a, p.118, CS 74205, 35042 Rennes, 13 France 14 15 16 Corresponding authors: Martin Lascoux ([email protected]), Marion Orsucci 17 ([email protected]) 18 19 20 Running title: Influence of mating system on life history traits in Capsella spp. 21 22 23 Key words: mating system, ploidy, life history traits, environmental disturbance 24 25 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.26.398016; this version posted November 27, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 26 SUMMARY 27 Self-fertilization has recurrently evolved from outcrossing. Self-fertilization provides an advantage 28 in the short-term as individuals do not require a mate to reproduce, but self-fertilization is also 29 associated with both decreased genetic diversity and accumulation of weakly deleterious mutations, 30 which could, however, be alleviated in polyploid selfers. -

Evolution and Diversification of the Plant Gibberellin Receptor GID1
Evolution and diversification of the plant gibberellin receptor GID1 Hideki Yoshidaa,b, Eiichi Tanimotoc, Takaaki Hiraia, Yohei Miyanoirid,e, Rie Mitania, Mayuko Kawamuraa, Mitsuhiro Takedad,f, Sayaka Takeharaa, Ko Hiranoa, Masatsune Kainoshod,g, Takashi Akagih, Makoto Matsuokaa,1, and Miyako Ueguchi-Tanakaa,1 aBioscience and Biotechnology Center, Nagoya University, Nagoya, 464-8601 Aichi, Japan; bKihara Institute for Biological Research, Yokohama City University, Yokohama, 244-0813 Kanagawa, Japan; cGraduate School of Natural Sciences, Nagoya City University, Nagoya, 467-8501 Aichi, Japan; dStructural Biology Research Center, Graduate School of Science, Nagoya University, Nagoya, 464-8601 Aichi, Japan; eResearch Center for State-of-the-Art Functional Protein Analysis, Institute for Protein Research, Osaka University, Suita, 565-0871 Osaka, Japan; fDepartment of Structural BioImaging, Faculty of Life Sciences, Kumamoto University, 862-0973 Kumamoto, Japan; gGraduate School of Science and Engineering, Tokyo Metropolitan University, Hachioji, 192-0397 Tokyo, Japan; and hGraduate School of Agriculture, Kyoto University, 606-8502 Kyoto, Japan Edited by Mark Estelle, University of California, San Diego, La Jolla, CA, and approved July 10, 2018 (received for review April 9, 2018) The plant gibberellin (GA) receptor GID1 shows sequence similarity erwort Marchantia polymorpha (5–7). Furthermore, Hirano et al. to carboxylesterase (CXE). Here, we report the molecular evolution (5) reported that GID1s in the lycophyte Selaginella moellen- of GID1 from establishment to functionally diverse forms in dorffii (SmGID1s) have unique properties in comparison with eudicots. By introducing 18 mutagenized rice GID1s into a rice angiosperm GID1s: namely, lower affinity to bioactive GAs and gid1 null mutant, we identified the amino acids crucial for higher affinity to inactive GAs (lower specificity). -

IEG News June Edition
June 2019 IEG News June edition News from the Head of the Department I wish everyone at the department a nice summer - relaxing vacations, and successful work in the field or elsewhere. Thank you all for contributions during the past academic year! For the upcoming fall, don’t forget the following important events: - The dean of biology has invited all senior scientists (“PIs”) to strategic discussions about biology at Uppsala University on August 28 - see mail sent by Staffan Svärd on June 5 for details and reg- istration. - The annual biology teacher days, August 22-23 - see mail from Henning Blom on June 3 for de- tails and registration. - The annual IEG Day will take place 8th of November - a full day of information, interactions, and discussion on topics of importance to the success of IEG as an excellent academic environment. There will also be research presentations, and a party. Old colleague moves on to new challenges Stefan Bertilsson leaves us for a professor position at the Department of Aquatic Sciences and As- sessment at SLU. Good luck to Stefan in his new job, and we look forward to coming inter-univer- sity cooperation! At the farewell celebration, we equipped him with some relevant tools for his fu- ture at the agricultural university. Stefan will still be around to some extent to finalize supervision. Photos courtesy of Lars Tranvik News from the administration Travelling on a business trip? Business trips must be booked through Lingmerth’s travel agency, which is the university’s pro- cured supplier, read more on the Employee Portal (Medarbetarportalen). -
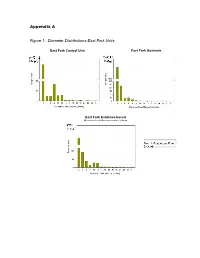
Appendix a Figure 1: Diameter Distributions-East Fork Units
Appendix A Figure 1: Diameter Distributions-East Fork Units East Fork Control Unit East Fork Goshawk Guidelines Unit East Fork Evidence-based Ecological Restoration Unit Figure 2: Diameter Distributions-Redondo Units Redondo Control Unit Redondo Goshawk Guidelines Unit Redondo Evidence-based Ecological Restoration Unit Figure 3: FlamMap Simulated Crown Fire Behavior: East Fork Project Area 20 mph Southwest Wind 25 mph 30 mph 35 mph 40 mph Map Legend Crown Fire Activity N No crown fire Passive crown activity fire (torching) Active crown fire (spreading) Important Note: Expected Crown Fire Behavior by Percent The 97th percentile of low fuel of Project Area moistures for June from 34 No Crown Passive years of data from the Kaibab Fire Crown Fire Active National Forest, Arizona were used in all simulations. This Windspeed Activity (torching) Crown Fire 20 79.9 0.9 19.2 input represents potential 25 71.2 1.9 26.9 extremes of fire weather during the peak of the fire 30 64.9 2.9 32.3 season, not average fire 35 57.3 4.7 38.0 season conditions. 40 48.7 9.4 42.0 Figure 4: FlamMap Simulated Crown Fire Behavior: Redondo Project Area 20 mph Southwest Wind 25 mph 30 mph 35 mph 40 mph Map Legend Crown Fire Activity N Highway 4 Valles Caldera No crown fire Passive crown activity entrance road fire (torching) Active crown fire (spreading) Important Note: Expected Crown Fire Behavior by Percent The 97th percentile of low fuel of Project Area moistures for June from 34 Passive years of data from the Kaibab No Crown Crown Fire Active Crown National Forest, Arizona were Windspeed Fire Activity (torching) Fire used in all simulations. -

Capsella Bursa-Pastoris ) – Establishment of a New Model System
Characterisation of the natural homeotic variety Stamenoid petals (Spe ) in the Shepherd´s Purse ( Capsella bursa-pastoris ) – Establishment of a new model system Dissertation zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) vorgelegt dem Rat der Biologisch-Pharmazeutischen Fakultät der Friedrich-Schiller-Universität Jena von Diplom-Biologin Pia Nutt Geboren am 9. Juni 1973 in Paderborn Gutachter 1. Prof. Dr. Günter Theißen (Jena) 2. Prof. Dr. Ralf Oelmüller (Jena) 3. PD Dr. Stefan Gleissberg (Ohio, USA) Tag der öffentlichen Verteidigung: Donnerstag, den 18. Dezember 2008 Meinen Eltern und Jorge Table of contents Table of contents 1 Introduction …………………………………………………………………….. 3 1.1 About homeosis………………………………………………………….. 3 1.2 Developmental genetics of floral homeotic mutants ……………………. 5 1.3 The role of homeotic mutants in the evolution of flowers……………….. 7 1.4 A floral homeotic variant of C. bursa-pastoris helps investigating the evolutionary role of homeosis in plants………………………………….. 8 1.5 Capsella bursa-pastoris as a model species……………………………… 10 1.6 Aims of this work………………………………………………………… 11 2 Overview of the manuscripts …………………………………………………… 14 3 Manuscript I ……………………………………………………………………... 16 P. Nutt , J. Ziermann, M. Hintz, B. Neuffer, and G. Theißen (2006): Capsella as a model system to study the evolutionary relevance of floral homeotic mutants. Plant Systematics and Evolution 259, pp 217-235. 4 Manuscript II ……………………………………………………………………. 36 P. Nutt 1, Janine Ziermann 1 and G. Theißen (submitted to The Plant Cell on May 7, 2008) Ectopic expression and co-segregation of an AGAMOUS orthologue in Stamenoid petals , a natural homeotic floral variant of Capsella bursa-pastoris. ( 1 These authors contributed equally to this work) 5 Manuscript III …………………………………………………………………… 84 C. -

Washington Flora Checklist a Checklist of the Vascular Plants of Washington State Hosted by the University of Washington Herbarium
Washington Flora Checklist A checklist of the Vascular Plants of Washington State Hosted by the University of Washington Herbarium The Washington Flora Checklist aims to be a complete list of the native and naturalized vascular plants of Washington State, with current classifications, nomenclature and synonymy. The checklist currently contains 3,929 terminal taxa (species, subspecies, and varieties). Taxa included in the checklist: * Native taxa whether extant, extirpated, or extinct. * Exotic taxa that are naturalized, escaped from cultivation, or persisting wild. * Waifs (e.g., ballast plants, escaped crop plants) and other scarcely collected exotics. * Interspecific hybrids that are frequent or self-maintaining. * Some unnamed taxa in the process of being described. Family classifications follow APG IV for angiosperms, PPG I (J. Syst. Evol. 54:563?603. 2016.) for pteridophytes, and Christenhusz et al. (Phytotaxa 19:55?70. 2011.) for gymnosperms, with a few exceptions. Nomenclature and synonymy at the rank of genus and below follows the 2nd Edition of the Flora of the Pacific Northwest except where superceded by new information. Accepted names are indicated with blue font; synonyms with black font. Native species and infraspecies are marked with boldface font. Please note: This is a working checklist, continuously updated. Use it at your discretion. Created from the Washington Flora Checklist Database on September 17th, 2018 at 9:47pm PST. Available online at http://biology.burke.washington.edu/waflora/checklist.php Comments and questions should be addressed to the checklist administrators: David Giblin ([email protected]) Peter Zika ([email protected]) Suggested citation: Weinmann, F., P.F. Zika, D.E. Giblin, B. -

A Vascular Flora Inventory
A Vascular Flora Inventory Ottawa Sands Ottawa County Parks, Michigan September 2020 Prepared by William Martinus & Associates Financial assistance for this project was provided, in part, by the Coastal Management Program, Water Resources Division, Michigan Department of Environment, Great Lakes, and Energy, under the National Coastal Zone Management program, through a grant from the National Oceanic and Atmospheric Administration, U.S. Department of Commerce. The statements, findings, conclusions, and recommendations in this report are those of the Ottawa County Parks & Recreation Commission and do not necessarily reflect the views of the Michigan Department of Environment, Great Lakes, and Energy or the National Oceanic and Atmospheric Administration. 1 Table of Contents I. Introduction and Purpose 3 II. Overview 3 III. Plant Communities 4 IV. Endangered, Threatened, and Special Concern Species 5 V. Species Lists 7 VI. References 21 2 I. Introduction and Purpose Ottawa Sands, Ottawa County Parks, consists of 345 acres including an 80-acre inland lake, natural forests, coastal dunes, intermittent wetlands, inundated shrub swamp, and riparian marsh, shrub, and swamp communities. The eleven natural plant communities occurring on the site are listed along with hundreds of associated plant and animal species. - Ottawa Sands is located near the mouth of the Grand River in sections 17, 18 and 20 of Spring Lake Township, Ottawa County, in Western Michigan. - Property includes 5,585 feet of Grand River frontage. - A Floristic Quality Assessment demonstrates that a diverse and extremely high-quality plant component exists at Ottawa Sands. Purpose - To gain an understanding of the vegetative plant communities and flora of western Ottawa County and central west Michigan area. -

Karyotype Evolution in Apomictic Boechera and the Origin of the Aberrant Chromosomes
The Plant Journal (2015) 82, 785–793 doi: 10.1111/tpj.12849 Karyotype evolution in apomictic Boechera and the origin of the aberrant chromosomes Terezie Mandakov a1, M. Eric Schranz2, Timothy F. Sharbel3, Hans de Jong4 and Martin A. Lysak1,* 1CEITEC – Central European Institute of Technology, Masaryk University, Brno, CZ-62500 Czech Republic, 2Plant Systematics Group, Wageningen University (WU), Droevendaalsesteeg 1, Wageningen, 6708 PB The Netherlands, 3Apomixis Research Group, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Gatersleben, D-06466 Germany, and 4Laboratory of Genetics, Wageningen UR PSG, P.O. Box 16, Wageningen, 6700 AA The Netherlands Received 17 February 2015; revised 24 March 2015; accepted 1 April 2015; published online 10 April 2015. *For correspondence (e-mail [email protected]). SUMMARY Chromosome rearrangements may result in both decrease and increase of chromosome numbers. Here we have used comparative chromosome painting (CCP) to reconstruct the pathways of descending and ascend- ing dysploidy in the genus Boechera (tribe Boechereae, Brassicaceae). We describe the origin and structure of three Boechera genomes and establish the origin of the previously described aberrant Het and Del chro- mosomes found in Boechera apomicts with euploid (2n = 14) and aneuploid (2n = 15) chromosome number. CCP analysis allowed us to reconstruct the origin of seven chromosomes in sexual B. stricta and apomictic B. divaricarpa from the ancestral karyotype (n = 8) of Brassicaceae lineage I. Whereas three chromosomes (BS4, BS6, and BS7) retained their ancestral structure, five chromosomes were reshuffled by reciprocal translocations to form chromosomes BS1-BS3 and BS5. The reduction of the chromosome number (from x = 8tox= 7) was accomplished through the inactivation of a paleocentromere on chromosome BS5. -

Group Code Species Code Species Common Name Classification
group code species code species common name classification genome version source transcript version Source website EUD Aco Aquilegia coerulea columbine Eudicotyledons (Ranunculales) JGI v1 Phytozome 11 JGI v1.1 https://phytozome.jgi.doe.gov/pz/portal.html BRA Aly Arabidopsis lyrata lyrate rockcress Eudicotyledons (Rosids – Malvids – Brassicales) JGI v1 Phytozome 11 JGI V1.0 https://phytozome.jgi.doe.gov/pz/portal.html BRA Ath Arabidopsis thaliana (Columbia) thale cress Eudicotyledons (Rosids – Malvids – Brassicales) TAIR10 Araport Araport11 https://araport.org EUD Bvu Beta vulgaris sugar beet Eudicotyledons (Caryophyllales) RefBeet-1.2.2 Ensembl RefBeet-1.2.2 https://plants.ensembl.org/info/website/ftp/index.html BRA Bst Boechera stricta drummond's rockcress Eudicotyledons (Rosids – Malvids – Brassicales) JGI v1 Phytozome 10 JGI v1.3 https://phytozome.jgi.doe.gov/pz/portal.html BRA Bra Brassica rapa turnip mustard Eudicotyledons (Rosids – Malvids – Brassicales) JGI v1 Phytozome 10 JGI v1.2 https://phytozome.jgi.doe.gov/pz/portal.html BRA Cgr Capsella grandiflora Eudicotyledons (Rosids – Malvids – Brassicales) JGI v1 Phytozome 10 JGI v1.1 https://phytozome.jgi.doe.gov/pz/portal.html BRA Cru Capsella rubella red shepherd's purse Eudicotyledons (Rosids – Malvids – Brassicales) JGI v1 Phytozome 10 ASGPB v0.4 https://phytozome.jgi.doe.gov/pz/portal.html BRA Cpa Carica papaya papaya Eudicotyledons (Rosids – Malvids - Brassicales) JGI v1 Phytozome 11 JGI v1.0 https://phytozome.jgi.doe.gov/pz/portal.html MAL Ccl Citrus clementina clementine Eudicotyledons -

100 Years of Change in the Flora of the Carolinas
ASTERACEAE 224 Zinnia Linnaeus 1759 (Zinnia) A genus of about 17 species, herbs, of sw. North America south to South America. References: Smith in FNA (2006c); Cronquist (1980)=SE. 1 Achenes wingless; receptacular bracts (chaff) toothed or erose on the lip..............................................................Z. peruviana 1 Achenes winged; receptacular bracts (chaff) with a differentiated fimbriate lip........................................................Z. violacea * Zinnia peruviana (Linnaeus) Linnaeus, Zinnia. Cp (GA, NC, SC): disturbed areas; rare (commonly cultivated), introduced from the New World tropics. May-November. [= FNA, K, SE; ? Z. pauciflora Linnaeus – S] * Zinnia violacea Cavanilles, Garden Zinnia. Cp (GA, NC, SC): disturbed areas; rare (commonly cultivated), introduced from the New World tropics. May-November. [= FNA, K; ? Z. elegans Jacquin – S, SE] BALSAMINACEAE A. Richard 1822 (Touch-me-not Family) A family of 2 genera and 850-1000 species, primarily of the Old World tropics. References: Fischer in Kubitzki (2004). Impatiens Linnaeus (Jewelweed, Touch-me-not, Snapweed, Balsam) A genus of 850-1000 species, herbs and subshrubs, primarily tropical and north temperate Old World. References: Fischer in Kubitzki (2004). 1 Corolla purple, pink, or white; plants 3-6 (-8) dm tall; stems puberulent or glabrous; [cultivated alien, rarely escaped]. 2 Sepal spur strongly recurved; stems puberulent..............................................................................................I. balsamina 2 Sepal spur slightly -

The MADS-Box Transcription Factor PHERES1 Controls
SHORT REPORT The MADS-box transcription factor PHERES1 controls imprinting in the endosperm by binding to domesticated transposons Rita A Batista1,2, Jordi Moreno-Romero1,2†, Yichun Qiu1,2, Joram van Boven1,2, Juan Santos-Gonza´ lez1,2, Duarte D Figueiredo1,2‡, Claudia Ko¨ hler1,2* 1Department of Plant Biology, Uppsala BioCenter, Swedish University of Agricultural Sciences, Uppsala, Sweden; 2Linnean Centre for Plant Biology, Swedish University of Agricultural Sciences, Uppsala, Sweden Abstract MADS-box transcription factors (TFs) are ubiquitous in eukaryotic organisms and play major roles during plant development. Nevertheless, their function in seed development remains largely unknown. Here, we show that the imprinted Arabidopsis thaliana MADS-box TF PHERES1 (PHE1) is a master regulator of paternally expressed imprinted genes, as well as of non-imprinted key regulators of endosperm development. PHE1 binding sites show distinct epigenetic modifications on maternal and paternal alleles, correlating with parental-specific transcriptional *For correspondence: activity. Importantly, we show that the CArG-box-like DNA-binding motifs that are bound by PHE1 [email protected] have been distributed by RC/Helitron transposable elements. Our data provide an example of the Present address: †Centre for molecular domestication of these elements which, by distributing PHE1 binding sites throughout Research in Agricultural the genome, have facilitated the recruitment of crucial endosperm regulators into a single Genomics (CRAG), CSIC-IRTA- transcriptional network. UAB-UB, Campus UAB, Barcelona, Spain; ‡Institute for Biochemistry and Biology, University of Potsdam, Potsdam, Germany Introduction MADS-box transcription factors (TFs) are present in most eukaryotes, and are classified into two Competing interests: The groups: type I or SRF (Serum Response Factor)-like, and type II or MEF2 (Myocyte Enhancing Fac- authors declare that no tor2)-like (Gramzow and Theissen, 2010). -

A Comprehensive Analysis of RALF Proteins in Green Plants Suggests There Are Two Distinct Functional Groups
ORIGINAL RESEARCH published: 24 January 2017 doi: 10.3389/fpls.2017.00037 A Comprehensive Analysis of RALF Proteins in Green Plants Suggests There Are Two Distinct Functional Groups Liam Campbell and Simon R. Turner * Faculty of Biology, Medicine and Health, School of Biological Science, University of Manchester, Manchester, UK Rapid Alkalinization Factors (RALFs) are small, cysteine-rich peptides known to be involved in various aspects of plant development and growth. Although RALF peptides have been identified within many species, a single wide-ranging phylogenetic analysis of the family across the plant kingdom has not yet been undertaken. Here, we identified RALF proteins from 51 plant species that represent a variety of land plant lineages. The inferred evolutionary history of the 795 identified RALFs suggests that the family has diverged into four major clades. We found that much of the variation across the family exists within the mature peptide region, suggesting clade-specific functional Edited by: diversification. Clades I, II, and III contain the features that have been identified as Madelaine Elisabeth Bartlett, University of Massachusetts Amherst, important for RALF activity, including the RRXL cleavage site and the YISY motif required USA for receptor binding. In contrast, members of clades IV that represent a third of the total Reviewed by: dataset, is highly diverged and lacks these features that are typical of RALFs. Members of Tatiana Arias, The Corporation for Biological clade IV also exhibit distinct expression patterns and physico-chemical properties. These Research, Colombia differences suggest a functional divergence of clades and consequently, we propose Ive De Smet, that the peptides within clade IV are not true RALFs, but are more accurately described Flanders Institute for Biotechnology, Belgium as RALF-related peptides.