RNA-Bloom Enables Reference-Free and Reference-Guided Sequence Assembly for Single-Cell Transcriptomes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Hexadecimal Number System and Memory Addressing
C5537_App C_1107_03/16/2005 APPENDIX C The Hexadecimal Number System and Memory Addressing nderstanding the number system and the coding system that computers use to U store data and communicate with each other is fundamental to understanding how computers work. Early attempts to invent an electronic computing device met with disappointing results as long as inventors tried to use the decimal number sys- tem, with the digits 0–9. Then John Atanasoff proposed using a coding system that expressed everything in terms of different sequences of only two numerals: one repre- sented by the presence of a charge and one represented by the absence of a charge. The numbering system that can be supported by the expression of only two numerals is called base 2, or binary; it was invented by Ada Lovelace many years before, using the numerals 0 and 1. Under Atanasoff’s design, all numbers and other characters would be converted to this binary number system, and all storage, comparisons, and arithmetic would be done using it. Even today, this is one of the basic principles of computers. Every character or number entered into a computer is first converted into a series of 0s and 1s. Many coding schemes and techniques have been invented to manipulate these 0s and 1s, called bits for binary digits. The most widespread binary coding scheme for microcomputers, which is recog- nized as the microcomputer standard, is called ASCII (American Standard Code for Information Interchange). (Appendix B lists the binary code for the basic 127- character set.) In ASCII, each character is assigned an 8-bit code called a byte. -

Midterm-2020-Solution.Pdf
HONOR CODE Questions Sheet. A Lets C. [6 Points] 1. What type of address (heap,stack,static,code) does each value evaluate to Book1, Book1->name, Book1->author, &Book2? [4] 2. Will all of the print statements execute as expected? If NO, write print statement which will not execute as expected?[2] B. Mystery [8 Points] 3. When the above code executes, which line is modified? How many times? [2] 4. What is the value of register a6 at the end ? [2] 5. What is the value of register a4 at the end ? [2] 6. In one sentence what is this program calculating ? [2] C. C-to-RISC V Tree Search; Fill in the blanks below [12 points] D. RISCV - The MOD operation [8 points] 19. The data segment starts at address 0x10000000. What are the memory locations modified by this program and what are their values ? E Floating Point [8 points.] 20. What is the smallest nonzero positive value that can be represented? Write your answer as a numerical expression in the answer packet? [2] 21. Consider some positive normalized floating point number where p is represented as: What is the distance (i.e. the difference) between p and the next-largest number after p that can be represented? [2] 22. Now instead let p be a positive denormalized number described asp = 2y x 0.significand. What is the distance between p and the next largest number after p that can be represented? [2] 23. Sort the following minifloat numbers. [2] F. Numbers. [5] 24. What is the smallest number that this system can represent 6 digits (assume unsigned) ? [1] 25. -

2018-19 MAP 160 Byte File Layout Specifications
2018-19 MAP 160 Byte File Layout Specifications OVERVIEW: A) ISAC will provide an Eligibility Status File (ESF) record for each student to all schools listed as a college choice on the student’s Student Aid Report (SAR). The ESF records will be available daily as Record Type = 7. ESF records may be retrieved via the File Extraction option in MAP. B) Schools will transmit Payment Requests to ISAC via File Transfer Protocol (FTP) using the MAP 160byte layout and identify these with Record Type = 4. C) When payment requests are processed, ISAC will provide payment results to schools through the MAP system. The payment results records can be retrieved in the 160 byte format using the MAP Payment Results File Extraction Option. MAP results records have a Record Type = 5. The MAP Payment Results file contains some eligibility status data elements. Also, the same student record may appear on both the Payment Results and the Eligibility Status extract files. Schools may also use the Reports option in MAP to obtain payment results. D) To cancel Payment Requests, the school with the current Payment Results record on ISAC's Payment Database must transmit a matching record with MAP Payment Request Code = C, with the Requested Award Amount field equal to zero and the Enrollment Hours field equal to 0 along with other required data elements. These records must be transmitted to ISAC as Record Type = 4. E) Summary of Data Element Changes, revision (highlighted in grey) made to the 2018-19 layout. NONE 1 2018-19 MAP 160 Byte File Layout Specifications – 9/17 2018-19 MAP 160 Byte File Layout Specifications F) The following 160 byte record layout will be used for transmitting data between schools and ISAC. -

POINTER (IN C/C++) What Is a Pointer?
POINTER (IN C/C++) What is a pointer? Variable in a program is something with a name, the value of which can vary. The way the compiler and linker handles this is that it assigns a specific block of memory within the computer to hold the value of that variable. • The left side is the value in memory. • The right side is the address of that memory Dereferencing: • int bar = *foo_ptr; • *foo_ptr = 42; // set foo to 42 which is also effect bar = 42 • To dereference ted, go to memory address of 1776, the value contain in that is 25 which is what we need. Differences between & and * & is the reference operator and can be read as "address of“ * is the dereference operator and can be read as "value pointed by" A variable referenced with & can be dereferenced with *. • Andy = 25; • Ted = &andy; All expressions below are true: • andy == 25 // true • &andy == 1776 // true • ted == 1776 // true • *ted == 25 // true How to declare pointer? • Type + “*” + name of variable. • Example: int * number; • char * c; • • number or c is a variable is called a pointer variable How to use pointer? • int foo; • int *foo_ptr = &foo; • foo_ptr is declared as a pointer to int. We have initialized it to point to foo. • foo occupies some memory. Its location in memory is called its address. &foo is the address of foo Assignment and pointer: • int *foo_pr = 5; // wrong • int foo = 5; • int *foo_pr = &foo; // correct way Change the pointer to the next memory block: • int foo = 5; • int *foo_pr = &foo; • foo_pr ++; Pointer arithmetics • char *mychar; // sizeof 1 byte • short *myshort; // sizeof 2 bytes • long *mylong; // sizeof 4 byts • mychar++; // increase by 1 byte • myshort++; // increase by 2 bytes • mylong++; // increase by 4 bytes Increase pointer is different from increase the dereference • *P++; // unary operation: go to the address of the pointer then increase its address and return a value • (*P)++; // get the value from the address of p then increase the value by 1 Arrays: • int array[] = {45,46,47}; • we can call the first element in the array by saying: *array or array[0]. -

Bits and Bytes
BITS AND BYTES To understand how a computer works, you need to understand the BINARY SYSTEM. The binary system is a numbering system that uses only two digits—0 and 1. Although this may seem strange to humans, it fits the computer perfectly! A computer chip is made up of circuits. For each circuit, there are two possibilities: An electric current flows through the circuit (ON), or An electric current does not flow through the circuit (OFF) The number 1 represents an “on” circuit. The number 0 represents an “off” circuit. The two digits, 0 and 1, are called bits. The word bit comes from binary digit: Binary digit = bit Every time the computer “reads” an instruction, it translates that instruction into a series of bits (0’s and 1’s). In most computers every letter, number, and symbol is translated into eight bits, a combination of eight 0’s and 1’s. For example the letter A is translated into 01000001. The letter B is 01000010. Every single keystroke on the keyboard translates into a different combination of eight bits. A group of eight bits is called a byte. Therefore, a byte is a combination of eight 0’s and 1’s. Eight bits = 1 byte Capacity of computer memory, storage such as USB devices, DVD’s are measured in bytes. For example a Word file might be 35 KB while a picture taken by a digital camera might be 4.5 MG. Hard drives normally are measured in GB or TB: Kilobyte (KB) approximately 1,000 bytes MegaByte (MB) approximately 1,000,000 (million) bytes Gigabtye (GB) approximately 1,000,000,000 (billion) bytes Terabyte (TB) approximately 1,000,000,000,000 (trillion) bytes The binary code that computers use is called the ASCII (American Standard Code for Information Interchange) code. -

IEEE Standard 754 for Binary Floating-Point Arithmetic
Work in Progress: Lecture Notes on the Status of IEEE 754 October 1, 1997 3:36 am Lecture Notes on the Status of IEEE Standard 754 for Binary Floating-Point Arithmetic Prof. W. Kahan Elect. Eng. & Computer Science University of California Berkeley CA 94720-1776 Introduction: Twenty years ago anarchy threatened floating-point arithmetic. Over a dozen commercially significant arithmetics boasted diverse wordsizes, precisions, rounding procedures and over/underflow behaviors, and more were in the works. “Portable” software intended to reconcile that numerical diversity had become unbearably costly to develop. Thirteen years ago, when IEEE 754 became official, major microprocessor manufacturers had already adopted it despite the challenge it posed to implementors. With unprecedented altruism, hardware designers had risen to its challenge in the belief that they would ease and encourage a vast burgeoning of numerical software. They did succeed to a considerable extent. Anyway, rounding anomalies that preoccupied all of us in the 1970s afflict only CRAY X-MPs — J90s now. Now atrophy threatens features of IEEE 754 caught in a vicious circle: Those features lack support in programming languages and compilers, so those features are mishandled and/or practically unusable, so those features are little known and less in demand, and so those features lack support in programming languages and compilers. To help break that circle, those features are discussed in these notes under the following headings: Representable Numbers, Normal and Subnormal, Infinite -

Numerical Computing with IEEE Floating Point Arithmetic This Page Intentionally Left Blank Numerical Computing with IEEE Floating Point Arithmetic
Numerical Computing with IEEE Floating Point Arithmetic This page intentionally left blank Numerical Computing with IEEE Floating Point Arithmetic Including One Theorem, One Rule of Thumb, and One Hundred and One Exercises Michael L. Overton Courant Institute of Mathematical Sciences New York University New York, New York siam. Society for Industrial and Applied Mathematics Philadelphia Copyright © 2001 by the Society for Industrial and Applied Mathematics. 1098765432 All rights reserved. Printed in the United States of America. No part of this book may be reproduced, stored, or transmitted in any manner without the written permission of the publisher. For information, write to the Society for Industrial and Applied Mathematics, 3600 University City Science Center, Philadelphia, PA 19104-2688. Library of Congress Cataloging-in-Publication Data Overton, Michael L Numerical computing with IEEE floating point arithmetic / Michael L Overton. p. cm. Includes bibliographical references and index. ISBN 0-89871-571-7 I. Computer arithmetic. 2. Floating-point arithmetic. 3. Numerical calculations. I. Title. QA76.9.M35O94200I O04'.0l'5l--dc2l 00-067941 SlcLJTL is a registered trademark. Dedicated to girls who like math especially my daughter Eleuthera Overton Sa This page intentionally left blank Contents Preface ix Acknowledgments xi 1 Introduction 1 2 The Real Numbers 5 3 Computer Representation of Numbers 9 4 IEEE Floating Point Representation 17 5 Rounding 25 6 Correctly Rounded Floating Point Operations 31 7 Exceptions 41 8 The Intel Microprocessors 49 9 Programming Languages 55 10 Floating Point in C 59 11 Cancellation 71 12 Conditioning of Problems 77 13 Stability of Algorithms 83 14 Conclusion 97 Bibliography 101 vii This page intentionally left blank Preface Numerical computing is a vital part of the modern scientific infrastructure. -
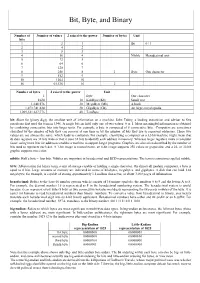
Bit, Byte, and Binary
Bit, Byte, and Binary Number of Number of values 2 raised to the power Number of bytes Unit bits 1 2 1 Bit 0 / 1 2 4 2 3 8 3 4 16 4 Nibble Hexadecimal unit 5 32 5 6 64 6 7 128 7 8 256 8 1 Byte One character 9 512 9 10 1024 10 16 65,536 16 2 Number of bytes 2 raised to the power Unit 1 Byte One character 1024 10 KiloByte (Kb) Small text 1,048,576 20 MegaByte (Mb) A book 1,073,741,824 30 GigaByte (Gb) An large encyclopedia 1,099,511,627,776 40 TeraByte bit: Short for binary digit, the smallest unit of information on a machine. John Tukey, a leading statistician and adviser to five presidents first used the term in 1946. A single bit can hold only one of two values: 0 or 1. More meaningful information is obtained by combining consecutive bits into larger units. For example, a byte is composed of 8 consecutive bits. Computers are sometimes classified by the number of bits they can process at one time or by the number of bits they use to represent addresses. These two values are not always the same, which leads to confusion. For example, classifying a computer as a 32-bit machine might mean that its data registers are 32 bits wide or that it uses 32 bits to identify each address in memory. Whereas larger registers make a computer faster, using more bits for addresses enables a machine to support larger programs. -

MIPS Floating Point
CS352H: Computer Systems Architecture Lecture 6: MIPS Floating Point September 17, 2009 University of Texas at Austin CS352H - Computer Systems Architecture Fall 2009 Don Fussell Floating Point Representation for dynamically rescalable numbers Including very small and very large numbers, non-integers Like scientific notation –2.34 × 1056 +0.002 × 10–4 normalized +987.02 × 109 not normalized In binary yyyy ±1.xxxxxxx2 × 2 Types float and double in C University of Texas at Austin CS352H - Computer Systems Architecture Fall 2009 Don Fussell 2 Floating Point Standard Defined by IEEE Std 754-1985 Developed in response to divergence of representations Portability issues for scientific code Now almost universally adopted Two representations Single precision (32-bit) Double precision (64-bit) University of Texas at Austin CS352H - Computer Systems Architecture Fall 2009 Don Fussell 3 IEEE Floating-Point Format single: 8 bits single: 23 bits double: 11 bits double: 52 bits S Exponent Fraction x = (!1)S "(1+Fraction)"2(Exponent!Bias) S: sign bit (0 ⇒ non-negative, 1 ⇒ negative) Normalize significand: 1.0 ≤ |significand| < 2.0 Always has a leading pre-binary-point 1 bit, so no need to represent it explicitly (hidden bit) Significand is Fraction with the “1.” restored Exponent: excess representation: actual exponent + Bias Ensures exponent is unsigned Single: Bias = 127; Double: Bias = 1203 University of Texas at Austin CS352H - Computer Systems Architecture Fall 2009 Don Fussell 4 Single-Precision Range Exponents 00000000 and 11111111 reserved Smallest -

Preprocessing C++ : Meta-Class Aspects
Preprocessing C++ : Meta-Class Aspects Edward D. Willink Racal Research Limited, Worton Drive, Reading, England +44 118 923 8278 [email protected] Vyacheslav B. Muchnick Department of Computing, University of Surrey, Guildford, England +44 1483 300800 x2206 [email protected] ABSTRACT C++ satisfies the previously conflicting goals of Object-Orientation and run-time efficiency within an industrial strength language. Run-time efficiency is achieved by ignoring the meta-level aspects of Object-Orientation. In a companion paper [15] we show how extensions that replace the traditional preprocessor lexical substitution by an Object-Oriented meta-level substitution fit naturally into C++. In this paper, we place those extensions in the context of a compile-time meta-level, in which application meta-programs can execute to browse, check or create program declarations. An extended example identifies the marshalling aspect as a programming concern that can be fully separated and automatically generated by an application meta-program. Keywords Object-Oriented Language; Preprocessor; C++; Meta-Level; Composition; Weaving; Aspect-Oriented Programming; Pattern Implementation 1 INTRODUCTION Prior to C++, Object Orientation, as exemplified by Smalltalk, was perceived to be inherently inefficient because of the run-time costs associated with message dispatch. C++ introduced a more restrictive Object Model that enabled most of the run-time costs to be resolved by compile-time computation. As a result Object Orientation in C++ is efficient and widely used. C++ requires that the layout of objects be frozen at compile-time, and that the type of the recipient of any message is known. The layout constraint enables a single contiguous memory allocation for each object. -

4 Data Types
4 Data Types VHDL is a strongly typed language. Every constant, signal, variable, function, and parameter is declared with a type, such as BOOLEAN or INTEGER, and can hold or return only a value of that type. VHDL predefines abstract data types, such as BOOLEAN, which are part of most programming languages, and hardware-re- lated types, such as BIT, found in most hardware languages. VHDL predefined types are declared in the STANDARD pack- age, which is supplied with all VHDL implementations (see Example 4–14). Data types addresses information about Enumeration Types Enumeration Overloading Enumeration Encoding Integer Types Array Types Record Types Predefined VHDL Data Types HOME CONTENTS INDEX For further assistance, email [email protected] or call your local support center V3.4 VHDL Compiler Reference Unsupported Data Types Synopsys Data Types Subtypes The advantage of strong typing is that VHDL tools can catch many common design errors, such as assigning an eight-bit value to a four-bit-wide signal, or incrementing an array index out of its range. The following code shows the definition of a new type, BYTE, as an array of eight bits, and a variable declaration, ADDEND, that uses this type. type BYTE is array(7 downto 0) of BIT; variable ADDEND: BYTE; The predefined VHDL data types are built from the basic VHDL data types. Some VHDL types are not supported for synthesis, such as REAL and FILE. The examples in this chapter show type definitions and associated object declarations. Although each constant, signal, variable, function, and parameter is declared with a type, only variable and signal declarations are shown here in the examples. -

Data Types in C
Princeton University Computer Science 217: Introduction to Programming Systems Data Types in C 1 Goals of C Designers wanted C to: But also: Support system programming Support application programming Be low-level Be portable Be easy for people to handle Be easy for computers to handle • Conflicting goals on multiple dimensions! • Result: different design decisions than Java 2 Primitive Data Types • integer data types • floating-point data types • pointer data types • no character data type (use small integer types instead) • no character string data type (use arrays of small ints instead) • no logical or boolean data types (use integers instead) For “under the hood” details, stay tuned for “number systems” lecture next week 3 Integer Data Types Integer types of various sizes: signed char, short, int, long • char is 1 byte • Number of bits per byte is unspecified! (but in the 21st century, pretty safe to assume it’s 8) • Sizes of other integer types not fully specified but constrained: • int was intended to be “natural word size” • 2 ≤ sizeof(short) ≤ sizeof(int) ≤ sizeof(long) On ArmLab: • Natural word size: 8 bytes (“64-bit machine”) • char: 1 byte • short: 2 bytes • int: 4 bytes (compatibility with widespread 32-bit code) 8 bytes • long: What decisions did the designers of Java make? 4 Integer Literals • Decimal: 123 • Octal: 0173 = 123 • Hexadecimal: 0x7B = 123 • Use "L" suffix to indicate long literal • No suffix to indicate short literal; instead must use cast Examples • int: 123, 0173, 0x7B • long: 123L, 0173L, 0x7BL • short: (short)123,