The Evolutionary History of Small Rnas in the Solanaceae
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Challenges to Genome Sequence Dissection in Sweetpotato
Breeding Science 67: 35–40 (2017) doi:10.1270/jsbbs.16186 Review Challenges to genome sequence dissection in sweetpotato Sachiko Isobe*, Kenta Shirasawa and Hideki Hirakawa Kazusa DNA Research Institute, Kazusa-Kamatari 2-6-7, Kisarazu, Chiba 292-0818, Japan The development of next generation sequencing (NGS) technologies has enabled the determination of whole genome sequences in many non-model plant species. However, genome sequencing in sweetpotato (Ipomoea batatas (L.) Lam) is still difficult because of the hexaploid genome structure. Previous studies suggested that a diploid wild relative, I. trifida (H.B.K.) Don., is the most possible ancestor of sweetpotato. Therefore, the ge- netic and genomic features of I. trifida have been studied as a potential reference for sweetpotato. Meanwhile, several research groups have begun the challenging task of directly sequencing the sweetpotato genome. In this manuscript, we review the recent results and activities of large-scale genome and transcriptome analysis related to genome sequence dissection in sweetpotato under the sections as follows: I. trifida genome and tran- script sequencing, genome sequences of I. nil (Japanese morning glory), transcript sequences in sweetpotato, chloroplast sequences, transposable elements and transfer DNA. The recent international activities of de novo whole genome sequencing in sweetpotato are also described. The large-scale publically available genome and transcript sequence resources and the international genome sequencing streams are expected to promote the genome sequence dissection in sweetpotato. Key Words: sweetpotato, Ipomoea, genome, transcript. Introduction tor diploid species are often sequenced and used as a reference for polyploidy species (Bertioli et al. 2016, The development of next generation sequencing (NGS) D’Hont et al. -

Evolutionary Routes to Biochemical Innovation Revealed by Integrative
RESEARCH ARTICLE Evolutionary routes to biochemical innovation revealed by integrative analysis of a plant-defense related specialized metabolic pathway Gaurav D Moghe1†, Bryan J Leong1,2, Steven M Hurney1,3, A Daniel Jones1,3, Robert L Last1,2* 1Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, United States; 2Department of Plant Biology, Michigan State University, East Lansing, United States; 3Department of Chemistry, Michigan State University, East Lansing, United States Abstract The diversity of life on Earth is a result of continual innovations in molecular networks influencing morphology and physiology. Plant specialized metabolism produces hundreds of thousands of compounds, offering striking examples of these innovations. To understand how this novelty is generated, we investigated the evolution of the Solanaceae family-specific, trichome- localized acylsugar biosynthetic pathway using a combination of mass spectrometry, RNA-seq, enzyme assays, RNAi and phylogenomics in different non-model species. Our results reveal hundreds of acylsugars produced across the Solanaceae family and even within a single plant, built on simple sugar cores. The relatively short biosynthetic pathway experienced repeated cycles of *For correspondence: [email protected] innovation over the last 100 million years that include gene duplication and divergence, gene loss, evolution of substrate preference and promiscuity. This study provides mechanistic insights into the † Present address: Section of emergence of plant chemical novelty, and offers a template for investigating the ~300,000 non- Plant Biology, School of model plant species that remain underexplored. Integrative Plant Sciences, DOI: https://doi.org/10.7554/eLife.28468.001 Cornell University, Ithaca, United States Competing interests: The authors declare that no Introduction competing interests exist. -

Sweet Potato (Ipomoea Batatas L
Cláudio E. Cartabiano-Leite, et. al. International Journal of Engineering Research and Applications www.ijera.com ISSN: 2248-9622, Vol. 10, Issue 6, (Series-VIII) June 2020, pp. 23-40 RESEARCH ARTICLE OPEN ACCESS Sweet potato (Ipomoea batatas L. Lam) nutritional potential and social relevance: a review Cláudio E. Cartabiano-Leite*, Ornella M. Porcu**, Alicia F. de Casas* * Department of Food Science and Technology, Federal University of Santa Catarina, Itacorubi, Florianópolis/SC, 88034-001, Brazil ** Department of Chemistry, Federal Technological University of Paraná, Via do Conhecimento, Km 1, Bloco V, Pato Branco/PR, 85503-390, Brazil Corresponding author: Cláudio E. Cartabiano-Leite ABSTRACT Sweet potato (Ipomoea batatas (L.) Lam) is a dicotyledonous angiosperm plant which belongs to the Convolvulaceae family and its capable of producing nutritious tuberous roots eaten worldwide. Its origin, as well as the circumstances related to its worldwide dispersion, are pertinent questions and intrigue researchers till nowadays. China is the main sweet potato producing country, and the Asian continent has the largest share of world production. In Brazil, sweet potatoes are specially grown by small farmers and used to domestic market supply. The sweet potato arouses huge interest when considering its nutritional qualities, mainly because it is rich in fibers, micronutrients, and an excellent source of energy for the consumer. The colored pulp cultivars such as yellow, orange, and purple sweet potatoes have in their composition several bioactive compounds such as polyphenols, carotenoids, and anthocyanins. In this regard, the work presents a review of the main aspects related to taxonomy, morphology, history, world production, and Brazilian production, highlighting the nutritional potential and the social relevance of sweet potatoes as a crop. -

Voluntary Guidelines for the Conservation and Sustainable Use of Crop Wild Relatives Food Plants
Voluntary Guidelines for the Conservation and Sustainable Use of Crop Wild Relatives Food Plants Crop wild relatives are potential sources of desirable traits for breeding well-adapted varieties while wild food plants constitute important components of the diets of many people worldwide. Voluntary Guidelines Unfortunately, their natural wild habitats are increasingly under threat from both human activities and natural disasters. Habitat for the Conservation loss has a direct, negative impact on the diversity of these valuable and Sustainable resources. These guidelines, intended as reference materials for preparing a National Plan for the Conservation and Sustainable Use of Crop Wild Use of Crop Wild Relatives and Wild Food Plants, will contribute to stemming this continuing loss in diversity. The guidelines are Relatives and Wild therefore a useful tool for development practitioners, researchers, students and policy-makers who work on the conservation and Food Plants sustainable use of these valuable resources. ISBN 978-92-5-109919-3 9 789251 099193 FAO I7788EN/1/09.17 Voluntary Guidelines for the Conservation and Sustainable Use of Crop Wild Relatives and Wild Food Plants Food and Agriculture Organization of the United Nations Rome, 2017 VOLUNTARY GUIDELINES FOR THE CONSERVATION AND SUSTAINABLE USE OF CROP WILD RELATIVES AND WILD FOOD PLANTS The designations employed and the presentation of material in this information product do not imply the expression of any opinion whatsoever on the part of the Food and Agriculture Organization of the United Nations (FAO) concerning the legal or development status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. -

Disentangling the Origins of Cultivated Sweet Potato (Ipomoea Batatas (L.) Lam.)
4/20/2017 Disentangling the Origins of Cultivated Sweet Potato (Ipomoea batatas (L.) Lam.) Disentangling the Origins of Cultivated Sweet Potato (Ipomoea batatas (L.) Lam.) Caroline Roullier , Anne Duputié, Paul Wennekes, Laure Benoit, Víctor Manuel Fernández Bringas, Genoveva Rossel, David Tay, Doyle McKey, Vincent Lebot Published: May 27, 2013 https://doi.org/10.1371/journal.pone.0062707 Correction 15 Oct 2013: Roullier C, Duputié A, Wennekes P, Benoit L, Fernández Bringas VM, et al. (2013) Correction: Disentangling the Origins of Cultivated Sweet Potato (Ipomoea batatas (L.) Lam.). PLOS ONE 8(10): 10.1371/annotation/936fe9b441cb494d 87a3a6d9a37c6c68. https://doi.org/10.1371/annotation/936fe9b441cb494d87a3a6d9a37c6c68 | View correction Abstract Sweet potato (Ipomoea batatas (L.) Lam., Convolvulaceae) counts among the most widely cultivated staple crops worldwide, yet the origins of its domestication remain unclear. This hexaploid species could have had either an autopolyploid origin, from the diploid I. trifida, or an allopolyploid origin, involving genomes of I. trifida and I. triloba. We generated molecular genetic data for a broad sample of cultivated sweet potatoes and its diploid and polyploid wild relatives, for noncoding chloroplast and nuclear ITS sequences, and nuclear SSRs. Our data did not support an allopolyploid origin for I. batatas, nor any contribution of I. triloba in the genome of domesticated sweet potato. I. trifida and I. batatas are closely related although they do not share haplotypes. Our data support an autopolyploid origin of sweet potato from the ancestor it shares with I. trifida, which might be similar to currently observed tetraploid wild Ipomoea accessions. -
![Comparative Rna-Seq Analysis of Phenotypically Different Sweet Potatoes (Ipomoea Batatas [L.] Lam)](https://docslib.b-cdn.net/cover/0936/comparative-rna-seq-analysis-of-phenotypically-different-sweet-potatoes-ipomoea-batatas-l-lam-3150936.webp)
Comparative Rna-Seq Analysis of Phenotypically Different Sweet Potatoes (Ipomoea Batatas [L.] Lam)
COMPARATIVE RNA-SEQ ANALYSIS OF PHENOTYPICALLY DIFFERENT SWEET POTATOES (IPOMOEA BATATAS [L.] LAM) by ELIZABETH FIEDLER A THESIS Submitted in partial fulfillment of the requirements for the degree of Master of Science in the Agricultural Sciences Graduate Program of Delaware State University DOVER, DELAWARE May 2019 This Thesis is approved by the following members of the Final Oral Review Committee: Dr. Venu (Kal) Kalavacharla, Committee Chairperson, Department of Agriculture and Natural Resources, Delaware State University Dr. Richard Barczewski, Committee Member, Department of Agriculture and Natural Resources, Delaware State University Dr. Marikis Alvarez, Committee Member, Department of Agriculture and Natural Resources, Delaware State University Dr. Muthusamy Manoharan, External Committee Member, Department of Agriculture, Fisheries and Human Sciences, University of Arkansas at Pine Bluff Comparative RNA-Seq Analysis of Phenotypically Different Sweet Potatoes (Ipomoea batatas [L.] Lam) Elizabeth Fiedler Faculty Advisor: Dr. Venu (Kal) Kalavacharla ABSTRACT Sweet potato is arguably one of Earth’s top ten most important crops. It is relatively low maintenance, packed with essential vitamins and nutrients, and in addition to serving as an effective food crop, it has been suggested for use as a material for synthesizing plastics and as a replacement for corn as a source for bioethanol production. Sweet potatoes are difficult to bring to seed, so most sweet potato plants are grown from slips, which are cuttings from sweet potato vines. This makes it very easy for sweet potato viruses to spread from generation to generation. Currently, virus disease complexes, which are infections of two or more viruses with a synergistic interaction, pose the biggest threat to sweet potato yields. -

J. Andrew Mcdonald Debra R. Hansen Joshua R. Mcdill Beryl B
A PHYLOGENETIC ASSESSMENT OF BREEDING SYSTEMS AND FLORAL MORPHOLOGY OF NORTH AMERICAN IPOMOEA (CONVOLVULACEAE) J. Andrew McDonald Debra R. Hansen Department of Biology Plant Resources Center and University of Texas – Pan American Section of Integrative Biology 1201 W. University Dr. 1 University Station F0404 Edinburg, Texas 78539, U.S.A. The University of Texas at Austin [email protected] Austin, Texas 78712, U.S.A. Joshua R. McDill Beryl B. Simpson Plant Resources Center and Plant Resources Center and Section of Integrative Biology Section of Integrative Biology 1 University Station F0404 1 University Station F0404 The University of Texas at Austin The University of Texas at Austin Austin, Texas 78712, U.S.A. Austin, Texas 78712, U.S.A. ABSTRACT A phylogenetic investigation of 68 species and two varieties of tropical and temperate North American Ipomoea (Convolvulaceae) using sequence data from the internal transcribed spacer region (ITS) with parsimony and Bayesian analyses revealed multiple origins of au- togamy. By assessing breeding systems and floral morphological characters in the context of this phylogeny, we estimate 16 independent origins of autogamy and 4 subsequent reversions to xenogamy. Transitions to autogamy are associated with reduced pollen-ovule ratios, decreased anther-stigma distance, and small flower size. Although the relationship between floral traits and breeding systems has been described in previous studies, this is the first investigation to examine this association in Ipomoea. RESUMEN Una investigación filogenética sobre 68 especies y dos variedades de Ipomoea (Convolvulaceae) en las zonas tropicales y templadas de Norteamérica, empleando datos de secuencias de ADN (ITS) con análisis de parsimonia y Bayesianos, demuestran orígenes múltiples de la autogamia. -
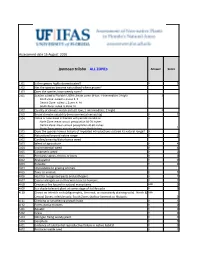
WRA.Datasheet.Template
Assessment date 16 August 2016 Ipomoea triloba ALL ZONEs Answer Score 1.01 Is the species highly domesticated? n 0 1.02 Has the species become naturalised where grown? 1.03 Does the species have weedy races? 2.01 Species suited to Florida's USDA climate zones (0-low; 1-intermediate; 2-high) 2 North Zone: suited to Zones 8, 9 Central Zone: suited to Zones 9, 10 South Zone: suited to Zone 10 2.02 Quality of climate match data (0-low; 1-intermediate; 2-high) 2 2.03 Broad climate suitability (environmental versatility) y 1 2.04 Native or naturalized in habitats with periodic inundation y North Zone: mean annual precipitation 50-70 inches Central Zone: mean annual precipitation 40-60 inches South Zone: mean annual precipitation 40-60 inches 1 2.05 Does the species have a history of repeated introductions outside its natural range? y 3.01 Naturalized beyond native range y 2 3.02 Garden/amenity/disturbance weed 3.03 Weed of agriculture y 4 3.04 Environmental weed y 4 3.05 Congeneric weed y 2 4.01 Produces spines, thorns or burrs n 0 4.02 Allelopathic n 0 4.03 Parasitic n 0 4.04 Unpalatable to grazing animals 4.05 Toxic to animals y 1 4.06 Host for recognised pests and pathogens y 1 4.07 Causes allergies or is otherwise toxic to humans y 1 4.08 Creates a fire hazard in natural ecosystems unk 0 4.09 Is a shade tolerant plant at some stage of its life cycle n 0 4.10 Grows on infertile soils (oligotrophic, limerock, or excessively draining soils). -

Linking the Potato Genome to the Conserved Ortholog
Lindqvist-Kreuze et al. BMC Genetics 2013, 14:51 http://www.biomedcentral.com/1471-2156/14/51 RESEARCH ARTICLE Open Access Linking the potato genome to the conserved ortholog set (COS) markers Hannele Lindqvist-Kreuze1*, Kwangsoo Cho1,4, Leticia Portal1, Flor Rodríguez1,2, Reinhard Simon1, Lukas A Mueller3, David M Spooner2 and Merideth Bonierbale1 Abstract Background: Conserved ortholog set (COS) markers are an important functional genomics resource that has greatly improved orthology detection in Asterid species. A comprehensive list of these markers is available at Sol Genomics Network (http://solgenomics.net/) and many of these have been placed on the genetic maps of a number of solanaceous species. Results: We amplified over 300 COS markers from eight potato accessions involving two diploid landraces of Solanum tuberosum Andigenum group (formerly classified as S. goniocalyx, S. phureja), and a dihaploid clone derived from a modern tetraploid cultivar of S. tuberosum and the wild species S. berthaultii, S. chomatophilum, and S. paucissectum. By BLASTn (Basic Local Alignment Search Tool of the NCBI, National Center for Biotechnology Information) algorithm we mapped the DNA sequences of these markers into the potato genome sequence. Additionally, we mapped a subset of these markers genetically in potato and present a comparison between the physical and genetic locations of these markers in potato and in comparison with the genetic location in tomato. We found that most of the COS markers are single-copy in the reference genome of potato and that the genetic location in tomato and physical location in potato sequence are mostly in agreement. However, we did find some COS markers that are present in multiple copies and those that map in unexpected locations. -

Catalogue of the Naturalized Flora of Taiwan
Taiwania, 49(1):16-31, 2004 Catalogue of the Naturalized Flora of Taiwan Shan-Huah Wu(1,3), Chang-Fu Hsieh(2), and Marcel Rejmánek(1) (Manuscript received 14 October, 2003; accepted 18 November, 2003) ABSTRACT: This study was conducted in years 2000 to 2003 to compile a comprehensive list of naturalized species with background information, including origins, life forms, habits, usages, year of the first collection, and status. Major herbaria (TAI, HAST, TAIF, and NCKU) were visited to examine specimen records. Relevant publications and reliable websites and many other resources were also examined extensively for background information. Total 341 species in 222 genera and 60 families were documented as naturalized in Taiwan, representing 7.9% of the local flora. Besides, 25 species were considered as possibly naturalized due to insufficient evidences. A large portion (90.6%) of species reported here, were reported as “weeds” in other countries. Almost a half of the naturalized species (48.3%) were introduced from Americas. KEYWORDS: Naturalized plant, Plant invasions, List,Taiwan. INTRODUCTION Despite the recent recognition of the impacts caused by invasive plants worldwide (Mooney and Hobbs, 2000; Vitousek et al., 1997), there are still many regions in the world where basic information on naturalized plant taxa and plant invasions is only anecdotal or completely lacking, e.g. Asia and neighboring regions (Corlett, 1988; Meyer, 2000; Turner, 1995; Enmoto, 1999; Pandey, 2000). In Taiwan, although occasional attention has been paid to the naturalized plants (Chen and Wu 1997; Peng et al. 1998a, 1998b; Peng and Yang, 1998; Chen et al., 1999; Kuoh and Chen, 2000; Chen and Wu, 2001; Yang, 2001; Yang and Peng, 2001), comprehensive studies on invasive species and plant invasions are still missing. -

ABSTRACT WINSLOW, BENJAMIN KENT. Interspecific Hybridization and Characterization of Variegation in Ornamental Sweetpotato
ABSTRACT WINSLOW, BENJAMIN KENT. Interspecific Hybridization and Characterization of Variegation in Ornamental Sweetpotato (Ipomoea batatas (L.) Lam.). (Under the direction of Dr. G. Craig Yencho). Sweetpotato (Ipomoea batatas (L.) Lam.) is an important food crop throughout the world and North Carolina is the top producer of sweetpotatoes in the US (NCDACS, 2011). The important contribution of sweetpotato to the global food production system is widely recognized. However, what is not generally recognized is that sweetpotatoes are also valued for their ornamental characteristics and ornamental sweetpotato (OSP) varieties have become an economically important crop in their own right. Novel OSP breeding, creating dramatic new plant types and leaf colors, has enabled sweetpotatoes to expand sales in the ornamental marketplace but there is considerable opportunity for the improvement of floral color and leaf variegation in this species. To improve these traits in future ornamental varieties, two separate breeding projects were conducted. The first breeding project was the interspecific hybridization of sweetpotato with morning glory species (Ipomoea L.). Since other species of Ipomoea have stunning floral diversity and other important ornamental traits, a series of interspecific crosses of sweetpotato with other Ipomoea species were conducted. Four hybrid families were produced with a total of 84 confirmed hybrid progeny. Sweetpotato crossed successfully with only two other closely related species, Ipomoea tabascana McDonald & Austin and I. trifida (Kunth) G.Don. Ipomoea tabascana was the most fecund paternal parent. The second breeding project involved the characterization of variegation in ornamental sweetpotato. Our large sweetpotato germplasm collection has over 100 variegated clones but only one of these variegated cultivars has been released. -

The Wild Sweetpotato (Ipomoea Trifida)
Li et al. BMC Plant Biology (2019) 19:119 https://doi.org/10.1186/s12870-019-1708-z RESEARCHARTICLE Open Access The wild sweetpotato (Ipomoea trifida) genome provides insights into storage root development Ming Li1,3*† , Songtao Yang2†, Wei Xu4†, Zhigang Pu1†, Junyan Feng1, Zhangying Wang5, Cong Zhang1, Meifang Peng1, Chunguang Du6, Feng Lin1, Changhe Wei3, Shuai Qiao2, Hongda Zou5, Lei Zhang4, Yan Li3, Huan Yang3, Anzhong Liao2, Wei Song2, Zhongren Zhang4,JiLi4, Kai Wang4, Yizheng Zhang3, Honghui Lin3*, Jinbo Zhang4* and Wenfang Tan2* Abstract Background: Sweetpotato (Ipomoea batatas (L.) Lam.) is the seventh most important crop in the world and is mainly cultivated for its underground storage root (SR). The genetic studies of this species have been hindered by a lack of high-quality reference sequence due to its complex genome structure. Diploid Ipomoea trifida is the closest relative and putative progenitor of sweetpotato, which is considered a model species for sweetpotato, including genetic, cytological, and physiological analyses. Results: Here, we generated the chromosome-scale genome sequence of SR-forming diploid I. trifida var. Y22 with high heterozygosity (2.20%). Although the chromosome-based synteny analysis revealed that the I. trifida shared conserved karyotype with Ipomoea nil after the separation, I. trifida had a much smaller genome than I. nil due to more efficient eliminations of LTR-retrotransposons and lack of species-specific amplification bursts of LTR-RTs. A comparison with four non-SR-forming species showed that the evolution of the beta-amylase gene family may be related to SR formation. We further investigated the relationship of the key gene BMY11 (with identity 47.12% to beta-amylase 1) with this important agronomic trait by both gene expression profiling and quantitative trait locus (QTL) mapping.