Thermodynamic Property Prediction for Solid Organic Compounds Based on Molecular Structure
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Standard Reference Materials
MBS 0F STAND PUBLICATIONS iffiJIimTk & TECH IIIIIII A 11 10 7 E57&&y NBS SPECIAL PUBLICATION 260-97 /?e AU o* U.S. DEPARTMENT OF COMMERCE/National Bureau of Standards Standard Reference Materials: Summary of the Coal, Ore, Mineral, Rock, and Refractory Standards Issued by the National Bureau of Standards 100 Radu Mavrodineanu and Thomas E. Gills • U57 260-97 1985 c2 . Tm he National Bureau of Standards was established by an act of Congress on March 3, 1901. The m Bureau's overall goal is to strengthen and advance the nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau's technical work is performed by the National Measurement Laboratory, the National Engineering Laboratory, the Institute for Computer Sciences and Technology, and the Institute for Materials Science and Engineering The National Measurement Laboratory 2 Provides the national system of physical and chemical measurement; • Basic Standards coordinates the system with measurement systems of other nations and • Radiation Research furnishes essential services leading to accurate and uniform physical and • Chemical Physics chemical measurement throughout the Nation's scientific community, in- • Analytical Chemistry dustry, and commerce; provides advisory -

University of São Paulo Sâopaulo - Bha7il Isrp-4 São Paulo 1988
& 20 ISRP-4 SÃO PAULO 1988 BOOK OF ABSTRACTS OCTOBER 3 - 7,1988 UNIVERSITY OF SÃO PAULO SÂOPAULO - BHA7IL ISRP-4 SÃO PAULO 1988 UNIVERSIDADE DE SÃO PAULO OCTOBER 3-7,1988 4th International Symposium on Radiation Physics BOOK OF ABSTRACTS Universidade de São Paulo Conselho Nacional de Desenvolvimento Científico e Tecnológico Financiadora de Estudos e Projetos Secretaria de Ciência e Tecnologia do Estado de São Paulo Comissão Nacional de Energia Nuclear ill Centro Latino-Americano de Física FAPESP Fundação de Amparo ã Pesquisa do Estado de São Paulo FOREWORD The 4th International Symposium on Radiation Physics - ISRP-4 was organized with the objective of gathering the physics radiation community in a meeting where the nost recent advances in this broad interdisciplinary area of science could be discussed. A big effort was made announcing the Symposium through mailing lists and journals. The response of the community was very good. A large number of abstracts were submitted and 212 papers were accepted. The Organizing Committee made a big effort to raise funds to subsidize the Conference, and the Brazilian funding institutions were very receptive in supporting the event. We are very grateful to all of them wich on, listed in the front page. unfortunately, the cost of travel represented a big obstacle for the participation of many scientists in the meeting, in particular for those from developing countries. Contrary to our expectations we had to retype all abstracts received. This was done in a very short time and so we apologize for any mistakes that could have remained. The Organizing Committee wants to thank the Secretaries that participated in the making of this book and in the other activities of the Symposium. -
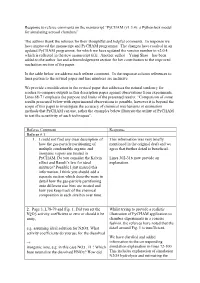
Response to Referee Comments on the Manuscript “Pycham (V1.3.4): a Python Box Model for Simulating Aerosol Chambers”
Response to referee comments on the manuscript “PyCHAM (v1.3.4): a Python box model for simulating aerosol chambers” The authors thank the referees for their thoughtful and helpful comments. In response we have improved the manuscript and PyCHAM programme. The changes have resulted in an updated PyCHAM programme, for which we have updated the version number to v2.0.4, which is reflected in the new manuscript title. Another author – Yunqi Shao – has been added to the author list and acknowledgement section for her contribution to the improved nucleation section of the paper. In the table below we address each referee comment. In the response column references to lines pertain to the revised paper and line numbers are inclusive. We provide a modification in the revised paper that addresses the natural tendency for readers to compare outputs in this description paper against observations from experiments. Lines 68-71 emphasis the purpose and limits of the presented results: “Comparison of some results presented below with experimental observations is possible, however it is beyond the scope of this paper to investigate the accuracy of chemical mechanisms or estimation methods that PyCHAM can use, rather the examples below illustrate the utility of PyCHAM to test the sensitivity of such techniques”. Referee Comment Response Referee # 1 1. I could not find any clear description of This information was very briefly how the gas-particle partitioning of mentioned in the original draft and we multiple condensable organic and agree that further detail is beneficial. inorganic vapors are treated in PyCHAM. Do you consider the Kelvin Lines 302-316 now provide an effect and Raoult’s law for ideal explanation. -

Theory and Applications of Fluctuating–Charge Models
THEORY AND APPLICATIONS OF FLUCTUATING–CHARGE MODELS BY JIAHAO CHEN B.S., University of Illinois at Urbana-Champaign, 2002 M.S., University of Illinois at Urbana-Champaign, 2008 DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemical Physics in the Graduate College of the University of Illinois at Urbana-Champaign, 2009 Urbana, Illinois Doctoral Committee: Professor Todd Martínez, Chair Professor Dirk Hundertmark Professor Duane Johnson Professor Nancy Makri Abstract Fluctuating–charge models are computationally efficient methods of treating polarization and charge–transfer phenomena in molecular mechanics and classical molecular dynamics simulations. They are also theoretically appealing as they are minimally parameterized, with parameters corresponding to the chemically important concepts of electronegativities and chemical hardness. However, they are known to overestimate charge transfer for widely separated atoms, leading to qualitative errors in the predicted charge distribution and exaggerated electrostatic properties. We present the charge transfer with polarization current equilibration (QTPIE) model, which solves this problem by introducing distance–dependent electronegativities. A graph–theoretic analysis of the topology of charge transfer allows us to relate the fundamental quantities of charge transfer back to the more familiar variables that represent atomic partial charges. This allows us to formulate a unified theoretical framework for fluctuating– charge models and topological charge descriptors. We also demonstrate the important role of charge screening effects in obtaining correct size extensivity in electrostatic properties. Analyzing the spatial symmetries of these properties allows us to shed light on the role of charge conservation in the electronegativity equalization process. Finally, we develop a water model for use in classical molecular dynamics simulations that is capable of treating both polarization and charge transfer phenomena. -

Article Partitioning and Gas–Wall Partitioning
Geosci. Model Dev., 14, 675–702, 2021 https://doi.org/10.5194/gmd-14-675-2021 © Author(s) 2021. This work is distributed under the Creative Commons Attribution 4.0 License. PyCHAM (v2.1.1): a Python box model for simulating aerosol chambers Simon Patrick O’Meara1,2, Shuxuan Xu1, David Topping1, M. Rami Alfarra1,2, Gerard Capes3, Douglas Lowe3, Yunqi Shao1, and Gordon McFiggans1 1Department for Earth and Environmental Sciences, University of Manchester, Manchester, M13 9PL, UK 2National Centre for Atmospheric Science, University of Manchester, Manchester, M13 9PL, UK 3Research Computing Services, University of Manchester, Manchester, M13 9PL, UK Correspondence: Gordon McFiggans (g.mcfi[email protected]) Received: 13 July 2020 – Discussion started: 20 August 2020 Revised: 2 November 2020 – Accepted: 10 December 2020 – Published: 2 February 2021 Abstract. In this paper the CHemistry with Aerosol Mi- 1 Introduction crophysics in Python (PyCHAM) box model software for aerosol chambers is described and assessed against bench- mark simulations for accuracy. The model solves the coupled Many major advances in atmospheric modelling have arisen system of ordinary differential equations for gas-phase chem- from chamber observations: for example, the partitioning of istry, gas–particle partitioning and gas–wall partitioning. Ad- vapours to particles (Odum et al., 1996), the gas-phase chem- ditionally, it can solve for coagulation, nucleation and parti- istry of ozone as part of the Master Chemical Mechanism cle loss to walls. PyCHAM is open-source, whilst the graph- (MCM) (Jenkin et al., 1997), and the gas-phase chemistries ical user interface, modular structure, manual, example plot- of limonene (Carslaw et al., 2012) and β-caryophyllene ting scripts, and suite of tests for troubleshooting and track- (Jenkin et al., 2012). -

In Silico Prediction of Physicochemical Properties
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by JRC Publications Repository In Silico Prediction of Physicochemical Properties a b John Dearden and Andrew Worth a QSTAR Consulting, Helsby, Cheshire, England b European Chemicals Bureau, Institute for Health & Consumer Protection, European Commission - Joint Research Centre, Ispra, Italy EUR 23051 EN - 2007 The mission of the IHCP is to provide scientific support to the development and implementation of EU policies related to health and consumer protection. The IHCP carries out research to improve the understanding of potential health risks posed by chemical, physical and biological agents from various sources to which consumers are exposed. European Commission Joint Research Centre Institute for Health and Consumer Protection Contact information Address: European Chemicals Bureau TP 581 E-mail: [email protected] Tel.: +39 0332 789566 Fax: +39 0332 786717 http:// http://ecb.jrc.it/QSAR http://www.jrc.cec.eu.int Legal Notice Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use which might be made of this publication. A great deal of additional information on the European Union is available on the Internet. It can be accessed through the Europa server http://europa.eu/ JRC 42061 EUR 23051 EN ISSN 1018-5593 Luxembourg: Office for Official Publications of the European Communities © European Communities, 2007 Reproduction is authorised provided the source is acknowledged Printed in Italy Quality control insert Name Signature Date Report Prepared Andrew Worth 03/12/07 by: Reviewed by: Tatiana Netzeva 10/12/07 (Scientific level) Approved by: Steven Eisenreich (Head of Unit) Final approval: Elke Anklam (IHCP Director) ABSTRACT This report provides a critical review of computational models, and in particular (quantitative) structure-property relationship (QSPR) models, that are available for the prediction of physicochemical properties. -

The Crystal Chemistry of Inorganic Hydroborates "2279
Review y The Crystal Chemistry of Inorganic Hydroborates Radovan Cernˇ ý * , Matteo Brighi and Fabrizio Murgia Laboratory of Crystallography, DQMP, University of Geneva, 24 quai E. Ansermet, CH-1211 Geneva, Switzerland; [email protected] (M.B.); [email protected] (F.M.) * Correspondence: [email protected] Dedicated to Dr. Howard Flack (1943–2017). y Academic Editor: Catherine Housecroft Received: 28 August 2020; Accepted: 23 September 2020; Published: 29 September 2020 Abstract: The crystal structures of inorganic hydroborates (salts and coordination compounds with anions containing hydrogen bonded to boron) except for the simplest anion, borohydride BH4−, are analyzed regarding their structural prototypes found in the inorganic databases such as Pearson’s Crystal Data [Villars and Cenzual (2015), Pearson’s Crystal Data. Crystal Structure Database for Inorganic Compounds, Release 2019/2020, ASM International, Materials Park, Ohio, USA]. Only the compounds with hydroborate as the only type of anion are reviewed, although including compounds gathering more than one different hydroborate (mixed anion). Carbaborane anions and partly halogenated hydroborates are included. Hydroborates containing anions other than hydroborate or neutral molecules such as NH3 are not discussed. The coordination polyhedra around the cations, including complex cations, and the hydroborate anions are determined and constitute the basis of the structural systematics underlying hydroborates chemistry in various variants of anionic packing. The latter is determined from anion–anion coordination with the help of topology analysis using the program TOPOS [Blatov (2006), IUCr CompComm. Newsl. 7, 4–38]. The Pauling rules for ionic crystals apply only to smaller cations with the observed coordination number within 2–4. -

Organic Component Vapor Pressures and Hygroscopicities of Aqueous Aerosol Measured by Optical Tweezers † ‡ ‡ ‡ † § § Chen Cai, , David J
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. Article pubs.acs.org/JPCA Organic Component Vapor Pressures and Hygroscopicities of Aqueous Aerosol Measured by Optical Tweezers † ‡ ‡ ‡ † § § Chen Cai, , David J. Stewart, Jonathan P. Reid,*, Yun-hong Zhang, Peter Ohm, Cari S. Dutcher, ∥ and Simon L. Clegg † The Institute of Chemical Physics, Key Laboratory of Cluster Science, Beijing Institute of Technology, Beijing 100081, People’s Republic of China ‡ School of Chemistry, University of Bristol, Bristol, BS8 1TS, U.K. § Department of Mechanical Engineering, University of Minnesota, 111 Church Street SE, Minneapolis, Minnesota 55455, United States ∥ School of Environmental Sciences, University of East Anglia, Norwich NR4 7TJ, U.K. *S Supporting Information ABSTRACT: Measurements of the hygroscopic response of aerosol and the particle-to-gas partitioning of semivolatile organic compounds are crucial for providing more accurate descriptions of the compositional and size distributions of atmospheric aerosol. Concurrent measurements of particle size and composition (inferred from refractive index) are reported here using optical tweezers to isolate and probe individual aerosol droplets over extended timeframes. The measurements are shown to allow accurate retrievals of component vapor pressures and hygroscopic response through examining corre- lated variations in size and composition for binary droplets containing water and a single organic component. Measurements are reported for a homologous series of dicarboxylic acids, maleic acid, citric acid, glycerol, or 1,2,6-hexanetriol. An assessment of the inherent uncertainties in such measurements when measuring only particle size is provided to confirm the value of such a correlational approach. -

Phd Dissertation De Zordi Nicola.Pdf
UNIVERSITÀ DEGLI STUDI DI TRIESTE XXIV CICLO DEL DOTTORATO DI RICERCA IN Scienze e Tecnologie Chimiche e Farmaceutiche MODIFIED RELEASE OF PHARMACEUTICAL DOSAGE FORMS Settore scientifico-disciplinare CHIM/09 DOTTORANDO NICOLA DE ZORDI COORDINATORE PROF. ENZO ALESSIO RELATORE / SUPERVISORE / TUTORE PROF. MARIAROSA MONEGHINI CORRELEATORE PROF. IRENEO KIKIC Solid dispersion : ANNO ACCADEMICO 2010/ 2011 Abstract 1 Solid dispersion : Abstract 2 Abstract During these three years, the research was focused on the preparation of pharmaceutical solid oral dosage forms with the aim to improve the dissolution behaviors and bioavailability of poor soluble drugs, or preparing sustained release systems for water- soluble drugs. In order to improve the bioavailability of poor soluble drugs, we adopted two strategies: 1) the micronization of the drug to increase their surface area, 2) preparing solid dispersions (SDs) using hydrophilic carrier. As known in the SDs the drug is dispersed or solubilized in an inert excipient or matrix where the active ingredient could exist in finely crystalline or amorphous state. When the system is exposed to aqueous media, the carrier dissolved and the drug is released as a very fine colloidal particles. This greatly reduction in particles size and the following surface area increase, results in an improvement of the dissolution rate. In addition to bioavailability enhancement, SDs systems were also directed towards the development of extender-release dosage forms using lipophilic carriers. For both the formulative approaches, we investigated the application of microwave (MW) and supercritical fluids (SCF) as preparative methods. In particular, MW ware employed for the preparation of solid dispersion either for immediate or sustained release of drugs, while SCF were investigated for the micronization and preparation of solid with the aim to prepare immediate release systems. -

(Ctls) for Chapter 24, Code of Miami-Dade County, Florida
DERM Technical Report: Development of Cleanup Target Levels (CTLs) for Chapter 24, Code of Miami-Dade County, Florida Prepared for the Department of Environmental Resources Management (DERM), Miami-Dade County, Florida by J. Keith Tolson, M.S. Kristen E. Jordan, Ph.D. Hugo G. Ochoa, D.V.M., Ph.D. Stephen M. Roberts, Ph.D. Center for Environmental & Human Toxicology University of Florida Gainesville, Florida October 20, 2000 DERM Technical Report – October 20, 2000 Table of Contents I. Introduction...........................................................................................................................5 II. Development of Groundwater CTLs.......................................................................................6 A. Introduction.....................................................................................................................6 B. Human Risk Equation.......................................................................................................7 1. Risk versus Hazard ....................................................................................................7 2. Exposure Parameters ...............................................................................................10 3. Toxicity Values ........................................................................................................10 III. Surface Water CTLs............................................................................................................14 A. Introduction...................................................................................................................14 -

To Go Get It> Now We Are Ready to Continue
First of all, be sure you have written or draw the triangle from the last page. If you haven't done so, <click here to go get it> Now we are ready to continue. If you want to go directly to a certain problem, click on the link below: Sample Problems: Solving for density Solving for volume Solving for mass Problem number one (solving for density): A piece of wood has a mass of 35 grams. If its volume is 7 cm3, What is its density? In order to solve this we have to do some different steps. First we write the formula as we intend to use it. Second, we place the numbers into the formula where they go (called the set- up) and lastly we do the mathematical calculations to attain the answer. Write the formula as we need to use it. In this question, they are Formula: Step One: asking for Density. So we are going to write the formula solving for that. D = m / v Now we have to figure out what value goes for each letter: the M D = 35g / Step Two: and the V. In this problem, it tells us the mass is 35g and the 3 volume is 7 cm3. So put them in the formula. 7cm Now the last and easiest part, do the math. Be sure you keep your Step Three: units with your problem. For example, in this one, its units will 5 g/cm3 be g/cm3 since that is what we did mathematically. <back to top> Problem number two (solving for volume): The density of a substance is 7.8 g/cm3. -

O-Nb(W) Systems That Exhibit Nonlinear Optical Properties
molecules Article New Solids in As-O-Mo, As(P)-O-Mo(W) and As(P)-O-Nb(W) Systems That Exhibit Nonlinear Optical Properties Nikolay Gerasimchuk 1,*, Lauri Kivijarvi 1, Bruce Noll 2, Meriem Goudjil 3, Soma Khanra 4, Yu Ping 4 , Miles Pearson 1 and Frank Röminger 5 1 Department of Chemistry, Missouri State University, Temple Hall 456, Springfield, MO 65897, USA; [email protected] (L.K.); [email protected] (M.P.) 2 Bruker AXS, Inc., Madison, WI 53722, USA; [email protected] 3 Laboratoire des Sciences des Matériaux, Faculté de Chimie, Université des Sciences et de la Technologie Houari Boumediène, Alger 16111, Algeria; [email protected] 4 Department of Physics and Astronomy, 220 Physics Building University of Missouri Columbia, Columbia, MO 65211, USA; [email protected] (S.K.); [email protected] (Y.P.) 5 Organisch-Chemisches Institut der Universität Heidelberg Lehrstuhl für Organische Chemie III 6 Im Neuenheimer Feld 270, Heidelberg University, D-69120 Heidelberg, Germany; [email protected] * Correspondence: [email protected]; Tel.: +1-(417)-836-5165 Abstract: Interactions between well-mixed fine powders of As2O3,P2O5, MoO3, WO3 and Nb2O5 at different stoichiometry in quartz ampoules under vacuum at ~1000 ◦C in the presence of metallic molybdenum (or niobium), over several weeks, led to shiny dichroic crystalline materials being formed in cooler parts of the reaction vessel. An addition of small quantities of metals-Mo or Citation: Gerasimchuk, N.; Kivijarvi, Nb-was made with the aim of partially reducing their highly oxidized Mo(VI), W(VI) or Nb(V) L.; Noll, B.; Goudjil, M.; Khanra, S.; species to corresponding Mo(V), W(V) and Nb(IV) centers, in order to form mixed valence solids.