2012) Rapid-Throughput Skeletal Phenotyping of 100 Knockout Mice Identifies 9 New Genes That Determine Bone Strength
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Clinicopathological and Molecular Genetic Analysis of Low-Grade Glioma in Adults
A CLINICOPATHOLOGICAL AND MOLECULAR GENETIC ANALYSIS OF LOW-GRADE GLIOMA IN ADULTS Presented by ANUSHREE SINGH MSc A thesis submitted in partial fulfilment of the requirements of the University of Wolverhampton for the degree of Doctor of Philosophy Brain Tumour Research Centre Research Institute in Healthcare Sciences Faculty of Science and Engineering University of Wolverhampton November 2014 i DECLARATION This work or any part thereof has not previously been presented in any form to the University or to any other body whether for the purposes of assessment, publication or for any other purpose (unless otherwise indicated). Save for any express acknowledgments, references and/or bibliographies cited in the work, I confirm that the intellectual content of the work is the result of my own efforts and of no other person. The right of Anushree Singh to be identified as author of this work is asserted in accordance with ss.77 and 78 of the Copyright, Designs and Patents Act 1988. At this date copyright is owned by the author. Signature: Anushree Date: 30th November 2014 ii ABSTRACT The aim of the study was to identify molecular markers that can determine progression of low grade glioma. This was done using various approaches such as IDH1 and IDH2 mutation analysis, MGMT methylation analysis, copy number analysis using array comparative genomic hybridisation and identification of differentially expressed miRNAs using miRNA microarray analysis. IDH1 mutation was present at a frequency of 71% in low grade glioma and was identified as an independent marker for improved OS in a multivariate analysis, which confirms the previous findings in low grade glioma studies. -

Genes in a Refined Smith-Magenis Syndrome Critical Deletion Interval on Chromosome 17P11.2 and the Syntenic Region of the Mouse
Downloaded from genome.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Article Genes in a Refined Smith-Magenis Syndrome Critical Deletion Interval on Chromosome 17p11.2 and the Syntenic Region of the Mouse Weimin Bi,1,6 Jiong Yan,1,6 Paweł Stankiewicz,1 Sung-Sup Park,1,7 Katherina Walz,1 Cornelius F. Boerkoel,1 Lorraine Potocki,1,3 Lisa G. Shaffer,1 Koen Devriendt,4 Małgorzata J.M. Nowaczyk,5 Ken Inoue,1 and James R. Lupski1,2,3,8 Departments of 1Molecular & Human Genetics, 2Pediatrics, Baylor College of Medicine, 3Texas Children’s Hospital, Houston, Texas 77030, USA; 4Centre for Human Genetics, University Hospital Gasthuisberg, Catholic University of Leuven, B-3000 Leuven, Belgium; 5Department of Pathology and Molecular Medicine, McMaster University, Hamilton, Ontario L8S 4J9, Canada Smith-Magenis syndrome (SMS) is a multiple congenital anomaly/mental retardation syndrome associated with behavioral abnormalities and sleep disturbance. Most patients have the same ∼4 Mb interstitial genomic deletion within chromosome 17p11.2. To investigate the molecular bases of the SMS phenotype, we constructed BAC/PAC contigs covering the SMS common deletion interval and its syntenic region on mouse chromosome 11. Comparative genome analysis reveals the absence of all three ∼200-kb SMS-REP low-copy repeats in the mouse and indicates that the evolution of SMS-REPs was accompanied by transposition of adjacent genes. Physical and genetic map comparisons in humans reveal reduced recombination in both sexes. Moreover, by examining the deleted regions in SMS patients with unusual-sized deletions, we refined the minimal Smith-Magenis critical region (SMCR) to an ∼1.1-Mb genomic interval that is syntenic to an ∼1.0-Mb region in the mouse. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

PRPSAP2 (NM 002767) Human Recombinant Protein – TP310719
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP310719 PRPSAP2 (NM_002767) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human phosphoribosyl pyrophosphate synthetase-associated protein 2 (PRPSAP2) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 40.7 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_002758 Locus ID: 5636 UniProt ID: O60256, A0A024QYY3 RefSeq Size: 2021 Cytogenetics: 17p11.2 RefSeq ORF: 1107 Synonyms: PAP41 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 PRPSAP2 (NM_002767) Human Recombinant Protein – TP310719 Summary: This gene encodes a protein that associates with the enzyme phosphoribosylpyrophosphate synthetase (PRS). PRS catalyzes the formation of phosphoribosylpyrophosphate which is a substrate for synthesis of purine and pyrimidine nucleotides, histidine, tryptophan and NAD. PRS exists as a complex with two catalytic subunits and two associated subunits. This gene encodes a non-catalytic associated subunit of PRS. -

Recombinant Human PRPSAP2 Protein Catalog Number: ATGP2461
Recombinant human PRPSAP2 protein Catalog Number: ATGP2461 PRODUCT INPORMATION Expression system E.coli Domain 1-369aa UniProt No. O60256 NCBI Accession No. NP_002758 Alternative Names phosphoribosyl pyrophosphate synthetase-associated protein 2, PAP41 PRODUCT SPECIFICATION Molecular Weight 43.3 kDa (392aa) confirmed by MALDI-TOF Concentration 0.25mg/ml (determined by Bradford assay) Formulation Liquid in. 20mM Tris-HCl buffer (pH 8.0) containing 0.15M NaCl, 20% glycerol, 1mM DTT Purity > 90% by SDS-PAGE Tag His-Tag Application SDS-PAGE Storage Condition Can be stored at +2C to +8C for 1 week. For long term storage, aliquot and store at -20C to -80C. Avoid repeated freezing and thawing cycles. BACKGROUND Description PRPSAP2 is a protein that associates with the enzyme phosphoribosylpyrophosphate synthetase (PRS). PRS catalyzes the formation of phosphoribosylpyrophosphate which is a substrate for synthesis of purine and pyrimidine nucleotides, histidine, tryptophan and NAD. PRS exists as a complex with two catalytic subunits and two associated subunits. This gene encodes a non-catalytic associated subunit of PRS. Alternate splicing results in multiple transcript variants. Recombinant human PRPSAP2 protein, fused to His-tag at N-terminus, was expressed in E. coli and purified by using conventional chromatography techniques. 1 Recombinant human PRPSAP2 protein Catalog Number: ATGP2461 Amino acid Sequence MGSSHHHHHH SSGLVPRGSH MGSMFCVTPP ELETKMNITK GGLVLFSANS NSSCMELSKK IAERLGVEMG KVQVYQEPNR ETRVQIQESV RGKDVFIIQT VSKDVNTTIM ELLIMVYACK TSCAKSIIGV IPYFPYSKQC KMRKRGSIVS KLLASMMCKA GLTHLITMDL HQKEIQGFFN IPVDNLRASP FLLQYIQEEI PDYRNAVIVA KSPASAKRAQ SFAERLRLGI AVIHGEAQDA ESDLVDGRHS PPMVRSVAAI HPSLEIPMLI PKEKPPITVV GDVGGRIAII VDDIIDDVDS FLAAAETLKE RGAYKIFVMA THGLLSSDAP RRIEESAIDE VVVTNTIPHE VQKLQCPKIK TVDISMILSE AIRRIHNGES MSYLFRNIGL DD General References Katashima,R., et al. (1998) Biochim. Biophys. Acta 1396 (3), 245-250 DATA SDS-PAGE 3ug by SDS-PAGE under reducing condition and visualized by coomassie blue stain. -

Amy Elizabeth Defnet Contact Information: [email protected] Degree and Date to Be Conferred: Ph.D
Targeting the Activator Protein-1 Complex to Inhibit Airway Smooth Muscle Cell Hyperproliferation in Asthma Item Type dissertation Authors Defnet, Amy Elizabeth Publication Date 2021 Abstract Hyperproliferation of airway smooth muscle (ASM) cells leads to increased ASM mass causing airway obstruction in inflammatory diseases such as asthma. Currently, there are no effective therapies to modulate ASM cell proliferation that contributes to ... Keywords Activator Protein-1; airway smooth muscle; retinoic acid; Airway Remodeling; Asthma; Protein Kinases; Transcription Factor AP-1; Tretinoin Download date 29/09/2021 14:19:54 Link to Item http://hdl.handle.net/10713/15769 Amy Elizabeth Defnet Contact Information: [email protected] Degree and Date to be Conferred: Ph.D. Pharmaceutical Sciences, May 2021 PROFESSIONAL OBJECTIVE My career objective is to work in academic institution where I can develop myself as an educator and researcher. My research employs a cross-disciplinary training regimen, including frequent opportunities for scientific/public speaking and inter-departmental engagement. In preparation for future teaching responsibilities, I have cultivated core pedagogical techniques through the JHU-UMB Collaborative Teaching Fellowship and Quality Matters Online Teaching Program. Additionally, participation in several societies and volunteer groups have helped cultivate my leadership and communication skills. EDUCATION University of Maryland, Baltimore 2016-present • Ph.D. Pharmaceutical Sciences, anticipated completion 2021 Fairleigh Dickinson University, Florham 2012-2016 • B.S. Biological Sciences with a Minor in Chemistry, 2016 RESEARCH Graduate Research Dr. Paul Shapiro and Dr. Maureen Kane, University of Maryland, Baltimore Fall 2016- Present This study hopes to overcome therapeutic limitations in asthma treatment that lead to bronchoconstriction and airway remodeling through evaluation of a novel function- selective ERK1/2 inhibitor and a RAR agonist. -

Prpsap2 (NM 057131) Rat Tagged ORF Clone – RR211579L3
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RR211579L3 Prpsap2 (NM_057131) Rat Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: Prpsap2 (NM_057131) Rat Tagged ORF Clone Tag: Myc-DDK Symbol: Prpsap2 Synonyms: Pap41 Vector: pLenti-C-Myc-DDK-P2A-Puro (PS100092) E. coli Selection: Chloramphenicol (34 ug/mL) Cell Selection: Puromycin ORF Nucleotide The ORF insert of this clone is exactly the same as(RR211579). Sequence: Restriction Sites: SgfI-MluI Cloning Scheme: ACCN: NM_057131 ORF Size: 1107 bp This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Prpsap2 (NM_057131) Rat Tagged ORF Clone – RR211579L3 OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_057131.1, NP_476472.1 RefSeq Size: 1612 bp RefSeq ORF: 1110 bp Locus ID: 117272 UniProt ID: O08618 Gene Summary: associates with the catalytic subunits and 39 kDa associated protein of the ribose-phosphate diphosphokinase enzyme complex [RGD, Feb 2006] This product is to be used for laboratory only. -

Rare Copy Number Variants Contribute to Congenital Left-Sided Heart Disease
Rare Copy Number Variants Contribute to Congenital Left-Sided Heart Disease Marc-Phillip Hitz1,2, Louis-Philippe Lemieux-Perreault3, Christian Marshall4, Yassamin Feroz-Zada3, Robbie Davies5, Shi Wei Yang1, Anath Christopher Lionel4, Guylaine D’Amours6, Emmanuelle Lemyre6, Rebecca Cullum7, Jean-Luc Bigras1, Maryse Thibeault1, Philippe Chetaille8, Alexandre Montpetit9, Paul Khairy3, Bert Overduin10, Sabine Klaassen11, Pamela Hoodless7, Mona Nemer12, Alexandre F. R. Stewart13, Cornelius Boerkoel14, Stephen W. Scherer4, Andrea Richter6, Marie-Pierre Dube´ 3, Gregor Andelfinger1* 1 Cardiovascular Genetics, Department of Pediatrics, Centre Hospitalier Universitaire Sainte Justine, Universite´ de Montre´al, Montre´al, Que´bec, Canada, 2 Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, United Kingdom, 3 Adult Congenital Heart Centre, Montreal Heart Institute, Universite´ de Montre´al, Montre´al, Que´bec, Canada, 4 The Centre for Applied Genomics and Program in Genetics and Genome Biology, The Hospital for Sick Children, Toronto, Ontario, Canada, 5 University of Ottawa Heart Institute, Ottawa, Ontario, Canada, 6 Service of Medical Genetics, Department of Pediatrics, Centre Hospitalier Universitaire Sainte Justine, Universite´ de Montre´al, Montre´al, Que´bec, Canada, 7 Terry Fox Laboratory, British Columbia Cancer Agency, Vancouver, British Columbia, Canada, 8 Cardiology Service, Centre Me`re- Enfants, Centre Hospitalier Universitaire de Que´bec, Universite´ de Laval, Que´bec City, Que´bec, Canada, 9 Genome Quebec Innovation -
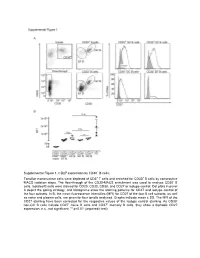
Supplemental Figure 1. CD27 Expression by CD30+ B Cells
Supplemental Figure 1. CD27 expression by CD30+ B cells. Tonsillar mononuclear cells were depleted of CD3+ T cells and enriched for CD30+ B cells by consecutive MACS isolation steps. The flow-through of the CD30-MACS enrichment was used to analyze CD30- B cells. Isolated B cells were stained for CD20, CD30, CD38, and CD27 or isotype control. Dot plots in panel A depict the gating strategy, and histograms show the staining patterns for CD27 and isotype control of the four subsets. In B, the mean fluorescence intensities (MFI) for CD27 of the four B cell subsets, as well as naive and plasma cells, are given for four tonsils analyzed. Graphs indicate mean ± SD. The MFI of the CD27 staining have been corrected for the respective values of the isotype control staining. As CD30- non-GC B cells include CD27- naïve B cells and CD27+ memory B cells, they show a biphasic CD27 expression. n.s., not significant; ** p<0.01 (unpaired t test). Supplemental Figure 2. Interleukin receptor expression by CD30+ B cells and relatedness of CD30+ EF B cells to CD21low B cells. mRNA and surface protein expression of IL2RB (A), IL21R (B) and CD21 (C) are shown for indicated B cell subsets of five tonsils (conventional (conv.) CD30- GC (germinal center) B cells (CD20highCD38+), CD30+ GC B cells, CD30- memory and CD30+ EF (extra follicular) B cells (CD20+CD38-/lowCD27+), and plasma cells (PC) (CD20+CD38high). mRNA data originate from Affymetrix genechip analyses (Tiacci et al., Blood, 2012; 120:4609-4620) and were analyzed using an unpaired t test. -

Accurate Prediction of Kinase-Substrate Networks Using
bioRxiv preprint doi: https://doi.org/10.1101/865055; this version posted December 4, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Accurate Prediction of Kinase-Substrate Networks Using Knowledge Graphs V´ıtNov´aˇcek1∗+, Gavin McGauran3, David Matallanas3, Adri´anVallejo Blanco3,4, Piero Conca2, Emir Mu~noz1,2, Luca Costabello2, Kamalesh Kanakaraj1, Zeeshan Nawaz1, Sameh K. Mohamed1, Pierre-Yves Vandenbussche2, Colm Ryan3, Walter Kolch3,5,6, Dirk Fey3,6∗ 1Data Science Institute, National University of Ireland Galway, Ireland 2Fujitsu Ireland Ltd., Co. Dublin, Ireland 3Systems Biology Ireland, University College Dublin, Belfield, Dublin 4, Ireland 4Department of Oncology, Universidad de Navarra, Pamplona, Spain 5Conway Institute of Biomolecular & Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland 6School of Medicine, University College Dublin, Belfield, Dublin 4, Ireland ∗ Corresponding authors ([email protected], [email protected]). + Lead author. 1 bioRxiv preprint doi: https://doi.org/10.1101/865055; this version posted December 4, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Phosphorylation of specific substrates by protein kinases is a key control mechanism for vital cell-fate decisions and other cellular pro- cesses. However, discovering specific kinase-substrate relationships is time-consuming and often rather serendipitous. -

Characterization of the Regulation of the Er Stress Response by the Dna Repair Enzyme Aag
DECIPHERING THE CROSSTALK: CHARACTERIZATION OF THE REGULATION OF THE ER STRESS RESPONSE BY THE DNA REPAIR ENZYME AAG Clara Forrer Charlier Faculty of Health and Medical Sciences Department of Biochemistry and Physiology This thesis is submitted for the degree of Doctor of Philosophy June 2018 DECIPHERING THE CROSSTALK: CHARACTERIZATION OF THE REGULATION OF THE ER STRESS RESPONSE BY THE DNA REPAIR ENZYME AAG- Clara Forrer Charlier – June 2018 SUMMARY The genome is a very dynamic store of genetic information and constantly threatened by endogenous and exogenous damaging agents. To maintain fidelity of the information stored, several robust and overlapping repair pathways, such as the Base Excision Repair (BER) pathway, have evolved. The main BER glycosylase responsible for repairing alkylation DNA damage is the alkyladenine DNA glycosylase (AAG). Repair initiated by AAG can lead to accumulation of cytotoxic intermediates. Here, we report the involvement of AAG in the elicitation of the unfolded protein response (UPR), a mechanism triggered to restore proteostasis in the cell whose dysfunction is implicated in diseases like diabetes, Alzheimer’s disease and cancer. Firstly, we determined that not only human ARPE-19 cells were capable of eliciting the UPR, but that an alkylating agent, methyl methanesulfonate (MMS), also triggers the response, and that in the absence of AAG the response is greatly diminished. Our luciferase reporter assay indicates that the response is activated on multiple branches (IRE1 and ATF6) on both AAG-proficient and deficient cells. Although no transcriptional induction of UPR markers was detected by RT-qPCR at 6 hours post MMS treatment, preliminary western-blot data at 6 and 24h, show activation of key UPR markers (p-eIF2α, BiP and XBP-1) upon MMS treatment in wild-type cells and little or no activation on AAG -/-. -

Rapid Phenotyping of Knockout Mice to Identify Genetic Determinants of Bone Strength
231 1 B FREUDENTHAL and others Rapid skeletal phenotyping of 231:1 R31–R46 Review knockout mice Open Access Rapid phenotyping of knockout mice to identify genetic determinants of bone strength Correspondence should be addressed Bernard Freudenthal1, John Logan1, Sanger Institute Mouse Pipelines2, to J H D Bassett or Peter I Croucher3, Graham R Williams1 and J H Duncan Bassett1 G R Williams Email 1Molecular Endocrinology Laboratory, Department of Medicine, Imperial College London, London, UK [email protected] or 2Mouse Pipelines, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK graham.williams@imperial. 3Garvan Institute of Medical Research, Sydney, New South Wales, Australia ac.uk Abstract The genetic determinants of osteoporosis remain poorly understood, and there is a Key Words large unmet need for new treatments in our ageing society. Thus, new approaches f osteoporosis for gene discovery in skeletal disease are required to complement the current f bone genome-wide association studies in human populations. The International Knockout f genetics Mouse Consortium (IKMC) and the International Mouse Phenotyping Consortium (IMPC) f gene discovery provide such an opportunity. The IKMC generates knockout mice representing each of Endocrinology of the known protein-coding genes in C57BL/6 mice and, as part of the IMPC initiative, the Origins of Bone and Cartilage Disease project identifies mutants with significant outlier skeletal phenotypes. This initiative will add value to data from large human cohorts and Journal provide a new understanding of bone and cartilage pathophysiology, ultimately leading to the identification of novel drug targets for the treatment of skeletal disease.