DISCOVERY and CHARACTERIZATION of LONG NON-CODING Rnas in PROSTATE CANCER by John R. Prensner a Dissertation Submitted in Parti
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Emergence of Lncrnas in Cancer Biology
Review The Emergence of lncRNAs in Cancer Biology John R. Prensner1,2 and Arul M. Chinnaiyan1–5 AbstAt R c The discovery of numerous noncoding RNA (ncRNA) transcripts in species from yeast to mammals has dramatically altered our understanding of cell biology, especially the biology of diseases such as cancer. In humans, the identification of abundant long ncRNA (lncRNA) >200 bp has catalyzed their characterization as critical components of cancer biology. Recently, roles for lncRNAs as drivers of tumor suppressive and oncogenic functions have appeared in prevalent cancer types, such as breast and prostate cancer. In this review, we highlight the emerging impact of ncRNAs in cancer research, with a particular focus on the mechanisms and functions of lncRNAs. Significance: lncRNAs represent the leading edge of cancer research. Their identity, function, and dysregulation in cancer are only beginning to be understood, and recent data suggest that they may serve as master drivers of carcinogenesis. Increased research on these RNAs will lead to a greater un- derstanding of cancer cell function and may lead to novel clinical applications in oncology. Cancer Discovery; 1(5):391–407. ©2011 AACR. intRoduction transcription reflects true biology or by-products of a leaky transcriptional system. Encompassed within these studies are A central question in biology has been, Which regions of the broad questions of what constitutes a human gene, what the human genome constitute its functional elements: those distinguishes a gene from a region that is simply transcribed, expressed as genes or those serving as regulatory elements? and how we interpret the biologic meaning of transcription. -

Molecular Distinctions Between Pediatric and Adult Mature B-Cell Non-Hodgkin Lymphomas Identified Through Genomic Profiling
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Public Health Resources Public Health Resources 4-19-2012 Molecular distinctions between pediatric and adult mature B-cell non-Hodgkin lymphomas identified through genomic profiling Karen Deffenbacher University of Nebraska Medical Center, Omaha Javeed Iqbal University of Nebraska Medical Center, Omaha Warren Sanger University of Nebraska Medical Center, Omaha Yulei Shen University of Nebraska Medical Center, Omaha Cynthia Lachel University of Nebraska Medical Center, Omaha See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/publichealthresources Part of the Public Health Commons Deffenbacher, Karen; Iqbal, Javeed; Sanger, Warren; Shen, Yulei; Lachel, Cynthia; Liu, Zhongfeng; Liu, Yanyan; Lim, Megan; Perkins, Sherrie; Fu, Kai; Smith, Lynette; Lynch, James; Staudt, Louis; Rimsza, Lisa M.; Jaffe, Elaine; Rosenwald, Andreas; Ott, German; Delabie, Jan; Campo, Elias; Gascoyne, Randy; Cairo, Mitchell; Weisenburger, Dennis; Greiner, Timothy; Gross, Thomas; and Chan, Wing, "Molecular distinctions between pediatric and adult mature B-cell non-Hodgkin lymphomas identified through genomic profiling" (2012). Public Health Resources. 145. https://digitalcommons.unl.edu/publichealthresources/145 This Article is brought to you for free and open access by the Public Health Resources at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Public Health Resources by an authorized administrator -

Expression and Functional Role of a Transcribed Noncoding RNA with an Ultraconserved Element in Hepatocellular Carcinoma
Expression and functional role of a transcribed noncoding RNA with an ultraconserved element in hepatocellular carcinoma Chiara Braconia, Nicola Valerib, Takayuki Kogurea, Pierluigi Gasparinib, Nianyuan Huanga, Gerard J. Nuovoc, Luigi Terraccianod, Carlo M. Croceb, and Tushar Patela,1 Departments of aInternal Medicine, bMolecular Virology, Immunology, and Medical Genetics, and cPathology, College of Medicine, Ohio State University, Columbus, OH 43210; and dMolecular Pathology Division, Institute of Pathology, University Hospital Basel, 4003 Basel, Switzerland Edited by Raymond L. White, University of California at San Francisco, Emeryville, CA, and approved December 9, 2010 (received for review July 28, 2010) Although expression of non–protein-coding RNA (ncRNA) can be shown to be involved in the modulation of cell proliferation and altered in human cancers, their functional relevance is unknown. apoptosis (12–15). Recent studies revealing the presence of Ultraconserved regions are noncoding genomic segments that are several hundred long transcribed ncRNAs raise the possibility 100% conserved across humans, mice, and rats. Conservation of that many ncRNAs contributing to cancer remain to be discov- gene sequences across species may indicate an essential functional ered (16). Other than microRNAs, however, only a handful of role, and therefore we evaluated the expression of ultraconserved ncRNAs have been implicated in hepatocarcinogenesis (17–20). RNAs (ucRNA) in hepatocellular cancer (HCC). The global expres- For the most part, the function of these ncRNAs is unknown. sion of ucRNAs was analyzed with a custom microarray. Expression Sequence conservation across species has been postulated to in- was verified in cell lines by real-time PCR or in tissues by in situ dicate that a given ncRNA may have a cellular function (21, 22). -

(12) Patent Application Publication (10) Pub. No.: US 2012/0322864 A1 Rossi Et Al
US 2012O322864A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0322864 A1 Rossi et al. (43) Pub. Date: Dec. 20, 2012 (54) SUSTAINED POLYPEPTIDE EXPRESSION (60) Provisional application No. 61/325,003, filed on Apr. FROM SYNTHETIC, MODIFIED RNAS AND 16, 2010, provisional application No. 61/387,220, USES THEREOF filed on Sep. 28, 2010. Publication Classification (75) Inventors: Derrick Rossi, Roslindale, MA (US); (51) Int. Cl. Luigi Warren, Cambridge, MA (US) CI2N5/07 (2010.01) A6IR 48/00 (2006.01) (73) Assignee: IMMUNE DISEASE INSTITUTE, C7H 2L/02 (2006.01) INC., Boston, MA (US) (52) U.S. Cl. ...................... 514/44 R; 536/23.5; 435/377 (57) ABSTRACT (21) Appl. No.: 13/590,880 Described herein are synthetic, modified RNAs for changing the phenotype of a cell. Such as expressing a polypeptide or altering the developmental potential. Accordingly, provided (22) Filed: Aug. 21, 2012 herein are compositions, methods, and kits comprising Syn thetic, modified RNAs for changing the phenotype of a cellor Related U.S. Application Data cells. These methods, compositions, and kits comprising Syn thetic, modified RNAs can be used either to express a desired (63) Continuation of application No. 13/088,009, filed on protein in a cell or tissue, or to change the differentiated Apr. 15, 2011. phenotype of a cell to that of another, desired cell type. Patent Application Publication Dec. 20, 2012 Sheet 1 of 45 US 2012/0322864 A1 s se xxx S s s nnnn s e sit. occa S. s Ra w s e s S. iiii is is iiiiSE SEE SS Patent Application Publication Dec. -

Comparative Analysis of the Ubiquitin-Proteasome System in Homo Sapiens and Saccharomyces Cerevisiae
Comparative Analysis of the Ubiquitin-proteasome system in Homo sapiens and Saccharomyces cerevisiae Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Universität zu Köln vorgelegt von Hartmut Scheel aus Rheinbach Köln, 2005 Berichterstatter: Prof. Dr. R. Jürgen Dohmen Prof. Dr. Thomas Langer Dr. Kay Hofmann Tag der mündlichen Prüfung: 18.07.2005 Zusammenfassung I Zusammenfassung Das Ubiquitin-Proteasom System (UPS) stellt den wichtigsten Abbauweg für intrazelluläre Proteine in eukaryotischen Zellen dar. Das abzubauende Protein wird zunächst über eine Enzym-Kaskade mit einer kovalent gebundenen Ubiquitinkette markiert. Anschließend wird das konjugierte Substrat vom Proteasom erkannt und proteolytisch gespalten. Ubiquitin besitzt eine Reihe von Homologen, die ebenfalls posttranslational an Proteine gekoppelt werden können, wie z.B. SUMO und NEDD8. Die hierbei verwendeten Aktivierungs- und Konjugations-Kaskaden sind vollständig analog zu der des Ubiquitin- Systems. Es ist charakteristisch für das UPS, daß sich die Vielzahl der daran beteiligten Proteine aus nur wenigen Proteinfamilien rekrutiert, die durch gemeinsame, funktionale Homologiedomänen gekennzeichnet sind. Einige dieser funktionalen Domänen sind auch in den Modifikations-Systemen der Ubiquitin-Homologen zu finden, jedoch verfügen diese Systeme zusätzlich über spezifische Domänentypen. Homologiedomänen lassen sich als mathematische Modelle in Form von Domänen- deskriptoren (Profile) beschreiben. Diese Deskriptoren können wiederum dazu verwendet werden, mit Hilfe geeigneter Verfahren eine gegebene Proteinsequenz auf das Vorliegen von entsprechenden Homologiedomänen zu untersuchen. Da die im UPS involvierten Homologie- domänen fast ausschließlich auf dieses System und seine Analoga beschränkt sind, können domänen-spezifische Profile zur Katalogisierung der UPS-relevanten Proteine einer Spezies verwendet werden. Auf dieser Basis können dann die entsprechenden UPS-Repertoires verschiedener Spezies miteinander verglichen werden. -

Global Patterns of Changes in the Gene Expression Associated with Genesis of Cancer a Dissertation Submitted in Partial Fulfillm
Global Patterns Of Changes In The Gene Expression Associated With Genesis Of Cancer A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy at George Mason University By Ganiraju Manyam Master of Science IIIT-Hyderabad, 2004 Bachelor of Engineering Bharatiar University, 2002 Director: Dr. Ancha Baranova, Associate Professor Department of Molecular & Microbiology Fall Semester 2009 George Mason University Fairfax, VA Copyright: 2009 Ganiraju Manyam All Rights Reserved ii DEDICATION To my parents Pattabhi Ramanna and Veera Venkata Satyavathi who introduced me to the joy of learning. To friends, family and colleagues who have contributed in work, thought, and support to this project. iii ACKNOWLEDGEMENTS I would like to thank my advisor, Dr. Ancha Baranova, whose tolerance, patience, guidance and encouragement helped me throughout the study. This dissertation would not have been possible without her ever ending support. She is very sincere and generous with her knowledge, availability, compassion, wisdom and feedback. I would also like to thank Dr. Vikas Chandhoke for funding my research generously during my doctoral study at George Mason University. Special thanks go to Dr. Patrick Gillevet, Dr. Alessandro Giuliani, Dr. Maria Stepanova who devoted their time to provide me with their valuable contributions and guidance to formulate this project. Thanks to the faculty of Molecular and Micro Biology (MMB) department, Dr. Jim Willett and Dr. Monique Vanhoek in embedding valuable thoughts to this dissertation by being in my dissertation committee. I would also like to thank the present and previous doctoral program directors, Dr. Daniel Cox and Dr. Geraldine Grant, for facilitating, allowing, and encouraging me to work in this project. -
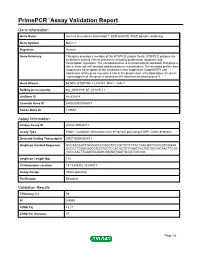
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name nucleus accumbens associated 1, BEN and BTB (POZ) domain containing Gene Symbol NACC1 Organism Human Gene Summary This gene encodes a member of the BTB/POZ protein family. BTB/POZ proteins are involved in several cellular processes including proliferation apoptosis and transcription regulation. The encoded protein is a transcriptional repressor that plays a role in stem cell self-renewal and pluripotency maintenance. The encoded protein also suppresses transcription of the candidate tumor suppressor Gadd45GIP1 and expression of this gene may play a role in the progression of multiple types of cancer. A pseudogene of this gene is located on the short arm of chromosome 9. Gene Aliases BEND8, BTBD14B, FLJ37383, NAC-1, NAC1 RefSeq Accession No. NC_000019.9, NT_011295.11 UniGene ID Hs.531614 Ensembl Gene ID ENSG00000160877 Entrez Gene ID 112939 Assay Information Unique Assay ID qHsaCIP0028717 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000292431 Amplicon Context Sequence GCCAACAGCTGCGGCACCGGCATCCGCTCTTCTACCAACGATCCCCGTCGGAA GCCCCTGGACAGCCGCGTGCTCCACGCTGTCAAGTACTACTGCCAGAACTTCGC CCCCAACTTCAAGGAGAGCGAGATGAATGCCATCGCGG Amplicon Length (bp) 115 Chromosome Location 19:13248302-13249017 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 98 R2 0.9995 cDNA Cq 19.17 cDNA Tm (Celsius) 87 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 27.88 Specificity (%) 100 Information -

Nac1 (NACC1) (NM 052876) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC208794 Nac1 (NACC1) (NM_052876) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: Nac1 (NACC1) (NM_052876) Human Tagged ORF Clone Tag: Myc-DDK Symbol: NACC1 Synonyms: BEND8; BTBD14B; BTBD30; NAC-1; NAC1; NECFM Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 4 Nac1 (NACC1) (NM_052876) Human Tagged ORF Clone – RC208794 ORF Nucleotide >RC208794 representing NM_052876 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGCCCAGACACTGCAGATGGAGATCCCGAACTTCGGCAACAGCATCCTGGAGTGCCTCAATGAACAGC GGCTGCAGGGCCTGTACTGTGACGTGTCAGTGGTGGTCAAGGGCCATGCCTTCAAGGCCCACCGGGCCGT GCTTGCTGCCAGCAGCTCCTACTTCCGGGACCTGTTCAACAACAGCCGCAGCGCCGTGGTGGAGCTGCCG GCGGCTGTGCAGCCCCAGTCTTTCCAGCAGATCCTCAGCTTCTGCTACACGGGCCGGCTGAGCATGAACG TGGGCGACCAGTTCCTGCTCATGTACACGGCTGGCTTCCTGCAGATCCAGGAGATCATGGAGAAGGGCAC CGAGTTCTTCCTCAAGGTGAGCTCCCCGAGCTGCGACTCCCAGGGCCTGCATGCGGAGGAGGCCCCATCG TCGGAGCCCCAGAGCCCCGTGGCGCAGACATCGGGCTGGCCAGCCTGTAGCACCCCGCTGCCCCTCGTGT CGCGGGTGAAGACGGAGCAGCAGGAGTCGGACTCCGTGCAGTGCATGCCCGTGGCCAAGCGGCTGTGGGA CAGTGGCCAGAAGGAGGCTGGGGGCGGCGGCAATGGCAGCCGCAAGATGGCCAAGTTCTCCACGCCGGAC -

Long Non-Coding Rnas Are Differentially Expressed in Hepatocellular Carcinoma Cell Lines with Different Metastatic Potential
DOI:http://dx.doi.org/10.7314/APJCP.2014.15.23.10513 Long non-coding RNAs are Differentially Expressed in Hepatocellular Carcinoma Cell Lines with Different Metastatic Potential RESEARCH ARTICLE Long Non-coding RNAs are Differentially Expressed in Hepatocellular Carcinoma Cell Lines with Differing Metastatic Potential Ting-Ting Fang, Xiao-Jing Sun, Jie Chen, Yan Zhao, Rui-Xia Sun, Ning Ren*, Bin-Bin Liu* Abstract Background: Metastasis is a major reason for poor prognosis in patients with cancer, including hepatocellular carcinoma (HCC). A salient feature is the ability of cancer cells to colonize different organs. Long non-coding RNAs (lncRNAs) play important roles in numerous cellular processes, including metastasis. Materials and Methods: In this study, the lncRNA expression profiles of two HCC cell lines, one with high potential for metastasis to the lung (HCCLM3) and the other to lymph nodes (HCCLYM-H2) were assessed using the Arraystar Human LncRNA Array v2.0, which contains 33,045 lncRNAs and 30,215 mRNAs. Coding-non-coding gene co-expression (CNC) networks were constructed and gene set enrichment analysis (GSEA) was performed to identify lncRNAs with potential functions in organ-specific metastasis. Levels of two representative lncRNAs and one representative mRNA, RP5-1014O16.1, lincRNA-TSPAN8 and TSPAN8, were further detected in HCC cell lines with differing metastasis potential by qRT-PCR. Results: Using microarray data, we identified 1,482 lncRNAs and 1,629 mRNAs that were differentially expressed (≥1.5 fold-change) between the two HCC cell lines. The most upregulated lncRNAs in H2 were RP11-672F9.1, RP5-1014O16.1, and RP11-501G6.1, while the most downregulated ones were lincRNA-TSPAN8, lincRNA-CALCA, C14orf132, NCRNA00173, and CR613944. -

Transcription and Chromatin Organization of a Housekeeping Gene Cluster Containing an Integrated B-Globin Locus Control Region
Transcription and Chromatin Organization of a Housekeeping Gene Cluster Containing an Integrated b-Globin Locus Control Region Daan Noordermeer1., Miguel R. Branco2., Erik Splinter1, Petra Klous1, Wilfred van IJcken3, Sigrid Swagemakers4, Manousos Koutsourakis1¤, Peter van der Spek4, Ana Pombo2*, Wouter de Laat1* 1 Department of Cell Biology and Genetics, Erasmus Medical Center, Rotterdam, The Netherlands, 2 MRC Clinical Sciences Centre, Faculty of Medicine, Imperial College London, London, United Kingdom, 3 Erasmus Center for Biomics, Erasmus Medical Center, Rotterdam, The Netherlands, 4 Erasmus Center for Bioinformatics, Erasmus Medical Center, Rotterdam, The Netherlands Abstract The activity of locus control regions (LCR) has been correlated with chromatin decondensation, spreading of active chromatin marks, locus repositioning away from its chromosome territory (CT), increased association with transcription factories, and long-range interactions via chromatin looping. To investigate the relative importance of these events in the regulation of gene expression, we targeted the human b-globin LCR in two opposite orientations to a gene-dense region in the mouse genome containing mostly housekeeping genes. We found that each oppositely oriented LCR influenced gene expression on both sides of the integration site and over a maximum distance of 150 kilobases. A subset of genes was transcriptionally enhanced, some of which in an LCR orientation-dependent manner. The locus resides mostly at the edge of its CT and integration of the LCR in either orientation caused a more frequent positioning of the locus away from its CT. Locus association with transcription factories increased moderately, both for loci at the edge and outside of the CT. These results show that nuclear repositioning is not sufficient to increase transcription of any given gene in this region. -

NAC1 Antibody (Human Preferred) A
Revision 1 C 0 2 - t NAC1 Antibody (Human Preferred) a e r o t S Orders: 877-616-CELL (2355) [email protected] Support: 877-678-TECH (8324) 0 2 Web: [email protected] 4 www.cellsignal.com 4 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source: UniProt ID: Entrez-Gene Id: WB H Endogenous 62 Rabbit Q96RE7 112939 Product Usage Information 7. Jinawath, N. et al. (2009) Oncogene 28, 1941-8. 8. Kim, J. et al. (2008) Cell 132, 1049-61. Application Dilution Western Blotting 1:1000 Storage Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA and 50% glycerol. Store at –20°C. Do not aliquot the antibody. Specificity / Sensitivity NAC1 Antibody (Human Preferred) detects endogenous levels of total NAC1 protein. Species Reactivity: Human Species predicted to react based on 100% sequence homology: Monkey Source / Purification Polyclonal antibodies are produced by immunizing animals with a synthetic peptide corresponding to an amino acid sequence at the carboxyl terminus of human NAC1. Antibodies are purified by protein A and peptide affinity chromatography. Background NAC1 or nuclear accumbens-1 is a nuclear factor that belongs to the POZ/BTB (Pox virus and zinc finger/bric-a-brac tramtrack broad complex) domain family. Also known as BTBD14B, it was originally identified in a unique neuronal forebrain structure responsible for reward motivation and addictive behaviors (1,2). NAC1 recruits HDAC3 and HDAC4 to transcriptionally repress gene expression in neuronal cells (3) and specifically co-represses other POZ/BTB proteins in the central nervous system (4). -

Individual Functions and Substrate Specificities of Importin Alpha
From the Max-Delbrück-Center for Molecular Medicine Berlin Head of the research group: Prof. Dr. M. Bader “Individual functions and substrate specificities of importin α subtypes” Dissertation for Fulfillment of Requirements for the Doctoral Degree of the University of Lübeck from the Department of Natural Sciences Submitted by Stefanie Hügel from Hennigsdorf Lübeck 2013 First referee: Prof. Dr. M. Bader Second referee: Prof. Dr. N. Tautz Date of oral examination: 21.02.13 Approved for printing. Lübeck, 22.02.2013 2 1 TABLE OF CONTENTS 1 TABLE OF CONTENTS _______________________________________________________ 3 2 ABSTRACT _______________________________________________________________ 6 3 ZUSAMMENFASSUNG ______________________________________________________ 7 4 INTRODUCTION ___________________________________________________________ 9 4.1 Nucleocytoplasmic protein transport ____________________________________________ 9 4.1.1 The nuclear pore complex – gateway to the nucleus ______________________________________ 10 4.1.1 Transport factors __________________________________________________________________ 11 4.1.2 Nothing happens without RanGTP _____________________________________________________ 12 4.2 The classical nuclear protein import pathway ____________________________________ 13 4.2.1 Structure and function of importin α ___________________________________________________ 13 4.2.2 Importin α:β mediated nuclear protein import___________________________________________ 15 4.2.3 The importin α protein family ________________________________________________________