Generation of Large Mitochondrial and Nuclear Nucleotide Sequences And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lord Howe Island Rodent Eradication Project NSW Species Impact Statement February 2017
Lord Howe Island Rodent Eradication Project NSW Species Impact Statement February 2017 Appendix K - Land Snail Survey 2016 K.1 Australian Museum Assessment of Potential Impacts on Land Snails Report Lord Howe Island Rodent Eradication Project: Assessment of potential impacts on land snails Frank Köhler1#, Isabel Hyman1, Adnan Moussalli2 1 – Australian Museum, Sydney, 2 – Museum Victoria, Melbourne, # - [email protected] 19 September 2016 Contents Summary ................................................................................................................................................. 2 Introduction: General characterisation of the land snail fauna ............................................................. 3 Diversity, endemism and distribution ................................................................................................. 3 Biology of the endemic species........................................................................................................... 6 General ecology of different land snail families ................................................................................. 7 Current status of the endangered species .......................................................................................... 9 Susceptibility to the baiting program ................................................................................................... 14 Toxicity of brodifacoum ................................................................................................................... -

Checklist of Fish and Invertebrates Listed in the CITES Appendices
JOINTS NATURE \=^ CONSERVATION COMMITTEE Checklist of fish and mvertebrates Usted in the CITES appendices JNCC REPORT (SSN0963-«OStl JOINT NATURE CONSERVATION COMMITTEE Report distribution Report Number: No. 238 Contract Number/JNCC project number: F7 1-12-332 Date received: 9 June 1995 Report tide: Checklist of fish and invertebrates listed in the CITES appendices Contract tide: Revised Checklists of CITES species database Contractor: World Conservation Monitoring Centre 219 Huntingdon Road, Cambridge, CB3 ODL Comments: A further fish and invertebrate edition in the Checklist series begun by NCC in 1979, revised and brought up to date with current CITES listings Restrictions: Distribution: JNCC report collection 2 copies Nature Conservancy Council for England, HQ, Library 1 copy Scottish Natural Heritage, HQ, Library 1 copy Countryside Council for Wales, HQ, Library 1 copy A T Smail, Copyright Libraries Agent, 100 Euston Road, London, NWl 2HQ 5 copies British Library, Legal Deposit Office, Boston Spa, Wetherby, West Yorkshire, LS23 7BQ 1 copy Chadwick-Healey Ltd, Cambridge Place, Cambridge, CB2 INR 1 copy BIOSIS UK, Garforth House, 54 Michlegate, York, YOl ILF 1 copy CITES Management and Scientific Authorities of EC Member States total 30 copies CITES Authorities, UK Dependencies total 13 copies CITES Secretariat 5 copies CITES Animals Committee chairman 1 copy European Commission DG Xl/D/2 1 copy World Conservation Monitoring Centre 20 copies TRAFFIC International 5 copies Animal Quarantine Station, Heathrow 1 copy Department of the Environment (GWD) 5 copies Foreign & Commonwealth Office (ESED) 1 copy HM Customs & Excise 3 copies M Bradley Taylor (ACPO) 1 copy ^\(\\ Joint Nature Conservation Committee Report No. -

Zoogeography of the Land and Fresh-Water Mollusca of the New Hebrides"
Web Moving Images Texts Audio Software Patron Info About IA Projects Home American Libraries | Canadian Libraries | Universal Library | Community Texts | Project Gutenberg | Children's Library | Biodiversity Heritage Library | Additional Collections Search: Texts Advanced Search Anonymous User (login or join us) Upload See other formats Full text of "Zoogeography of the land and fresh-water mollusca of the New Hebrides" LI E) RARY OF THE UNIVLRSITY Of ILLINOIS 590.5 FI V.43 cop. 3 NATURAL ri'^^OHY SURVEY. Zoogeography of the LAND AND FRESH-WATER MOLLUSCA OF THE New Hebrides ALAN SOLEM Curator, Division of Lower Invertebrates FIELDIANA: ZOOLOGY VOLUME 43, NUMBER 2 Published by CHICAGO NATURAL HISTORY MUSEUM OCTOBER 19, 1959 Library of Congress Catalog Card Number: 59-13761t PRINTED IN THE UNITED STATES OF AMERICA BY CHICAGO NATURAL HISTORY MUSEUM PRESS CONTENTS PAGE List of Illustrations 243 Introduction 245 Geology and Zoogeography 247 Phylogeny of the Land Snails 249 Age of the Land Mollusca 254 Land Snail Faunas of the Pacific Ocean Area 264 Land Snail Regions of the Indo-Pacific Area 305 converted by Web2PDFConvert.com Origin of the New Hebridean Fauna 311 Discussion 329 Conclusions 331 References 334 241 LIST OF ILLUSTRATIONS TEXT FIGURES PAGE 9. Proportionate representation of land snail orders in different faunas. ... 250 10. Phylogeny of land Mollusca 252 11. Phylogeny of Stylommatophora 253 12. Range of Streptaxidae, Corillidae, Caryodidae, Partulidae, and Assi- mineidae 266 13. Range of Punctinae, "Flammulinidae," and Tornatellinidae 267 14. Range of Clausiliidae, Pupinidae, and Helicinidae 268 15. Range of Bulimulidae, large Helicarionidae, and Microcystinae 269 16. Range of endemic Enidae, Cyclophoridae, Poteriidae, Achatinellidae and Amastridae 270 17. -

Entre Los Stylommatophora (Mollusca: Gastropoda)
Rev. peru. biol. 16(1): 051- 056 (Agosto 2009) © Facultad de Ciencias Biológicas UNMSM Posición evolutiva de BOSTRYX y SCUTALUS dentroVersión de Online los Stylommatophora ISSN 1727-9933 Posición evolutiva de caracoles terrestres peruanos (Orthalicidae) entre los Stylommatophora (Mollusca: Gastropoda) Evolutionary position of Peruvian land snails (Orthalicidae) among Stylommatophora (Mollusca: Gastropoda) Jorge Ramirez1,2, Rina Ramírez1,2, Pedro Romero1,2, Ana Chumbe1,2, Pablo Ramírez3 1Laboratorio de Sistemática Mole- cular y Filogeografía, Facultad de Resumen Ciencias Biológicas, Universidad Nacional Mayor de San Marcos. Los géneros Bostryx y Scutalus (Orthalicidae: Bulimulinae) son endémicos de América del Sur y están principal- Email Jorge Ramirez: jolobio@ mente distribuidos en la vertiente occidental de los Andes del Perú. El objetivo del presente trabajo fue evaluar hotmail.com su posición evolutiva dentro de los gastrópodos Stylommatophora basada en el marcador mitocondrial 16S 2Departamento de Malacología y Carcinología, Museo de Historia rRNA. Fueron obtenidas cuatro secuencias las que, junto con 28 de otros Stylommatophora disponibles en el Natural, Universidad Nacional GenBank, fueron alineadas con ClustalX. La reconstrucción filogenética se realizó mediante los métodos de Mayor de San Marcos. Neighbor-Joining, Máxima Parsimonia, Máxima Verosimilitud e Inferencia Bayesiana. El alineamiento resultó en Av. Arenales 1256, Apartado 14- 371 sitios, con presencia de indels. Los dos géneros de la Familia Orthalicidae por primera vez incluidos en una 0434, Lima-14, Perú. Email Rina filogenia molecular (Bostryx y Scutalus), formaron un grupo monofilético con otro miembro de la superfamilia Ramírez: [email protected] Orthalicoidea (Placostylus), tal como lo obtenido con marcadores nucleares. Se discute también su relación 3Laboratorio de Microbiología Molecular, Facultad de Ciencias evolutiva con otros caracoles terrestres. -

(Gastropoda, Euthyneura), I. Amphibuliminae
BASTERIA 37: 51-56, 1973 Catalogue of Bulimulidae (Gastropoda, Euthyneura), I. Amphibuliminae A.S.H. Breure Utrecht INTRODUCTION The Bulimulidae constitute a relatively large family, mainly confined South At 144 and to America. present the family includes genera number of subgenera. The specific and subspecific names available is estimated at about 3000. The subdivision of the family into Bulimulinae, Amphibuliminae, Odontostominae and Orthalicinae Placostylinae, is mainly based on shell features. This sensu lato conception of the Bulimulidae, already held by Pilsbry (1895-1902) and Thiele (1929-1931), is also favoured the by present author. More recent authors, e.g., Zilch (1959-1960), have accorded family rank to the subfamilies, the Placostylinae except- ed. However, the differences between the subfamilies are comparatively the differ from Bulimulinae slight, e.g., Amphibuliminae seem to the in the and the free muscle only palleal organs retractor system (Van Mol, 1971). in The Amphibuliminae are my opinion entirely confined to South A in which America. few African genera are included this subfamily, may better be placed elsewhere. The genus Aillya Odhner, 1928, occurring in Cameroon (West Africa), is placed here by Odhner (1928) on account of the anatomy. Baker (1955) placed the Aillyidae in the Heterurethra, near the Succineidae. Another African genus included in the Amphibuliminae is Prestonella Connolly, 1929. It occurs in South and is unknown. Africa its anatomy Some Asiatic species, referred to this subfamily, are also excluded from the present catalogue. 52 BASTERIA, Vol. 37, No. 3-4, 1973 The classification of the following Amphibuliminae is mainly ac- cording to Zilch (1959-1960): Simpulopsis (Simpulopsis) Beck, 1837. -

Review of the Fossil Record of the Australian Land Snail Genus
RECORDS OF THE WESTERN AUSTRALIAN MUSEUM 34 038–050 (2019) DOI: 10.18195/issn.0312-3162.34(1).2019.038-050 Review of the fossil record of the Australian land snail genus Bothriembryon Pilsbry, 1894 (Mollusca: Gastropoda: Bothriembryontidae): new distributional and geological data Corey S. Whisson1,2* and Helen E. Ryan3 1 Department of Aquatic Zoology, Western Australian Museum, Locked Bag 49, Welshpool DC, Western Australia 6986, Australia. 2 School of Veterinary and Life Sciences, Murdoch University, Murdoch, Western Australia 6150, Australia. 3 Department of Earth and Planetary Sciences, Western Australian Museum, Locked Bag 49, Welshpool DC, Western Australia 6986, Australia. * Corresponding author: [email protected] ABSTRACT – The land snail genus Bothriembryon Pilsbry, 1894, endemic to southern Australia, contains seven fossil and 39 extant species, and forms part of the Gondwanan family Bothriembryontidae. Little published data on the geographical distribution of fossil Bothriembryon exists. In this study, fossil and modern data of Bothriembryon from nine Australian museums and institutes were mapped for the first time. The fossilBothriembryon collection in the Western Australian Museum was curated to current taxonomy. Using this data set, the geological age of fossil and extant species was documented. Twenty two extant Bothriembryon species were identified in the fossil collection, with 15 of these species having a published fossil record for the first time. Several fossil and extant species had range extensions. The geological age span of Bothriembryon was determined as a minimum of Late Oligocene to recent, with extant endemic Western Australian Bothriembryon species determined as younger, traced to Pleistocene age. Extant Bothriembryon species from the Nullarbor region were older, dated Late Pliocene to Early Pleistocene. -

Placostylus Bivaricosus Breeding Project Final Report
Final Report Placostylus bivaricosus Breeding Project 13 December 2010 Ian Hutton PO Box 157 Lord Howe Island NSW 2898 Introduction to Placostylus bivaricosus The genus Placostylus is a group of large ground dwelling gastropods with a disjunct distribution in the Southwest Pacific from the Solomon Islands, Fiji and New Caledonia, to Lord Howe Island and the northern extremity of New Zealand. Three recent subspecies of the Lord Howe Island Placostylus are recognized. P. bivaricosus bivaricosus from the settlement area; P. bivaricosus cuniculinsulae from Blackburn Island (now presumed extinct) and P. bivaricosus etheridgeri from Little Slope considered likely to be extinct but there is a chance it may survive in isolated populations based on recent identification of hatchling snails from leaf litter collected on Little and Big Slopes in the 1970’s. The extant Lord Howe land snail Placostylus bivaricosus bivaricosus (from now on called Placostylus bivaricosus) is a large terrestrial snail found only on Lord Howe Island. This snail has a brown, pointed shell up to 7cm long and 2 cm in diameter. It is most closely related to New Zealand Placostylus species. Its status has declined from common to endangered. Historical accounts and fossil evidence indicate that the LHI Placostylus bivaricosus was formerly widespread and abundant on parts the island. The decline was first noted in the 1940’s and the species is now listed as critically endangered on the 2000 IUCN Red List of Threatened Species. The causes for decline of the LHI Placostylus are likely to include habitat clearing and modification, predation and habitat disturbance by introduced fauna (rats, Blackbirds and Song thrushes) and possibly herbicide and pesticide use. -

Proceedings of the United States National Museum
a Proceedings of the United States National Museum SMITHSONIAN INSTITUTION • WASHINGTON, D.C. Volume 121 1967 Number 3579 VALID ZOOLOGICAL NAMES OF THE PORTLAND CATALOGUE By Harald a. Rehder Research Curator, Division of Mollusks Introduction An outstanding patroness of the arts and sciences in eighteenth- century England was Lady Margaret Cavendish Bentinck, Duchess of Portland, wife of William, Second Duke of Portland. At Bulstrode in Buckinghamshire, magnificent summer residence of the Dukes of Portland, and in her London house in Whitehall, Lady Margaret— widow for the last 23 years of her life— entertained gentlemen in- terested in her extensive collection of natural history and objets d'art. Among these visitors were Sir Joseph Banks and Daniel Solander, pupil of Linnaeus. As her own particular interest was in conchology, she received from both of these men many specimens of shells gathered on Captain Cook's voyages. Apparently Solander spent considerable time working on the conchological collection, for his manuscript on descriptions of new shells was based largely on the "Portland Museum." When Lady Margaret died in 1785, her "Museum" was sold at auction. The task of preparing the collection for sale and compiling the sales catalogue fell to the Reverend John Lightfoot (1735-1788). For many years librarian and chaplain to the Duchess and scientif- 1 2 PROCEEDINGS OF THE NATIONAL MUSEUM vol. 121 ically inclined with a special leaning toward botany and conchology, he was well acquainted with the collection. It is not surprising he went to considerable trouble to give names and figure references to so many of the mollusks and other invertebrates that he listed. -

South Africa): Insights from Ecological and Genetic Studies
An investigation of the aquatic macroinvertebrate fauna of the southern Great Escarpment (South Africa): Insights from ecological and genetic studies A thesis submitted in fulfilment of the requirements of the degree of MASTER OF SCIENCE of RHODES UNIVERSITY by CHANTAL LEE TAYLOR JUNE 2015 Supervisor: Prof. N.P. Barker (Department of Botany, Rhodes University) Co-supervisor: Dr. H.M. Barber-James (Department of Freshwater Invertebrates, Albany Museum) Co-supervisor: Prof. M.H. Villet (Department of Entomology, Rhodes University) Abstract Biological diversity in freshwater biomes is vital to maintain healthy, functioning ecosystems with resilience to disturbance and the impacts of climate change. Freshwater ecosystems provide essential resources to life on Earth. However, as increasing pressure is being placed on the environment by human population growth, the quality of freshwater resources and the health of these ecosystems are at risk. Mountain streams provide an important source of water and are usually less affected by anthropogenic stressors, compared to lowland freshwaters. These montane streams are therefore of important conservation value and due to their untransformed nature serve as ideal ecosystems for biodiversity studies and as reference sites for studies on environmental change. This study explores aquatic macroinvertebrate biodiversity of the southern Great Escarpment in South Africa. Species assemblages and the environmental variables of each site were sampled from first order streams across five different mountain blocks along the Great Escarpment. Additionally, mitochondrial DNA of three mayfly species (Afroptilum sudafricanum, Demoreptus natalensis and Demoreptus capensis), commonly occurring in the study area, was analysed to compare the genetic diversity between habitat specialist and habitat generalist species. -

Integrated Conservation Approach for the Australian Land Snail Genus Bothriembryon Pilsbry, 1894: Curation, Taxonomy and Palaeontology
Integrated conservation approach for the Australian land snail genus Bothriembryon Pilsbry, 1894: curation, taxonomy and palaeontology Corey Whisson This thesis is presented for the Master of Philosophy 2019 Murdoch University, Perth, Western Australia DECLARATION I declare that this thesis is my own original account of my research and contains as its main content, work which has not previously been submitted for a degree at any tertiary education institution and as author, I reserve the right of copyright under the Copyright Act 1968, Commonwealth of Australia. …………………………………………………………… Corey Whisson i "Probably the most intriguing land shells in Australia are the bulimoid forms inhabiting the south-west corner. A large number of species and races has developed, and probably only a tithe has been described. It is unfortunate these have not yet been studied by anyone conversant with local conditions, and it is certain that they will provide future students with much research. No more exciting subject could be chosen by the student, but the unravelling of the many problems will necessitate much investigation." Mr Tom Iredale (cited in Iredale, 1939) ii Abstract Native land snails are important to ecosystems given their role in the decomposition process through herbivorous feeding of primarily decaying plant matter; calcium recycling and soil nitrification, and as a food source for larger predators. They also serve as a valuable bio-indicator group, especially in Western Australia where they are critical to Environmental Impact Assessments surveys. One conspicuous genus of native land snail found in Western Australia is Bothriembryon Pilsbry, 1894, a Gondwanan group endemic to the southern half of Australia and most diverse in the south-west of Western Australia. -

Exotic-Rodents-Background.Pdf
BACKGROUND DOCUMENT for the THREAT ABATEMENT PLAN to reduce the impacts of exotic rodents on biodiversity on Australian offshore islands of less than 100 000 hectares 2009 Contents 1 Introduction........................................................................................................................5 2 The problem with exotic rodents .....................................................................................6 2.1 Rodent species on Australian islands ........................................................................6 2.2 Australian islands with rodents...................................................................................7 2.3 Impacts of exotic rodents on island biodiversity and economic well-being.................8 3 Management of the threat...............................................................................................11 3.1 A response framework .............................................................................................11 3.2 Setting priorities........................................................................................................11 3.3 The toolbox...............................................................................................................12 3.4 Eradication ...............................................................................................................14 3.5 Sustained control......................................................................................................14 3.6 Stopping new invasions............................................................................................15 -
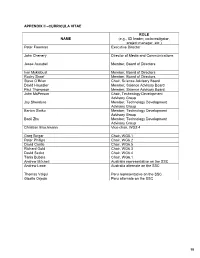
Appendix Ii –Curricula Vitae
APPENDIX II –CURRICULA VITAE ROL E NAME (e.g., ICI leader, co-investigator, project manager, etc.) Peter Freeman Executive Director John Chenery Director of Media and Communications Jesse Ausubel Member, Board of Directors Ivar Myklebust Member, Board of Directors Rocky Skeef Member, Board of Directors Steve O’Brien Chair, Science Advisory Board David Haussler Member, Science Advisory Board Paul Thompson Member, Science Advisory Board John McPerson Chair, Technology Development Advisory Group Jay Shendure Member, Technology Development Advisory Group Barton Slatko Member, Technology Development Advisory Group Baoli Zhu Member, Technology Development Advisory Group Christian Brochmann Vice-chair, WG2.4 Greg Singer Chair, WG5.1 Peter Phillips Chair, WG6.2 David Castle Chair, WG6.5 Richard Gold Chair, WG6.3 David Secko Chair, WG6.4 Tania Bubela Chair, WG6.1 Andrew Mitchell Australia representative on the SSC Andrew Lowe Australia alternate on the SSC Thomas Valqui Peru representative on the SSC Gisella Orjeda Peru alternate on the SSC 98 PETER FREEMAN BA, PHD, FIBD 120 Stuart Street, Guelph, Ontario, CANADA N1E 4S8 Phone (W): +1 519 824 4120 Cell: +1 519 731 2163 Email: [email protected] PROFILE Experienced science-based (PhD qualified) executive director, with a track record of successfully co- ordinating large multi-institutional research projects, networks and consortia in genomics, proteomics, stem cell research and population health. Strong financial management, strategic planning and project management skills gained in R&D and operations roles in the international malting and brewing industry Superior mentoring, facilitating, technical writing and editing skills used to develop successful interdisciplinary funding proposals. Fully proficient in information /communication technologies and their use in knowledge translation and public outreach activities.