Ionization and Dissociation Studies of Propadiene, 1, 3-Butadiene, 1,3
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trace Hydrocarbon Impurities in 1,3-Butadiene Using Optimized Rt®-Alumina BOND/MAPD PLOT Columns by Rick Morehead, Jan Pijpelink, Jaap De Zeeuw, Tom Vezza
Petroleum & Petrochemical Applications Analysis of Trace Hydrocarbon Impurities in 1,3-Butadiene Using Optimized Rt®-Alumina BOND/MAPD PLOT Columns By Rick Morehead, Jan Pijpelink, Jaap de Zeeuw, Tom Vezza Abstract Identifying and quantifying trace impurities in 1,3-butadiene is critical in producing high quality synthetic rubber products. Stan- dard analytical methods employ alumina PLOT columns which yield good resolution for low molecular weight hydrocarbons, but suffer from irreproducibility and poor sensitivity for polar hydrocarbons. In this study, Rt®-Alumina BOND/MAPD PLOT columns were used to separate both common light polar contaminants, including methyl acetylene and propadiene, as well as 4-vinylcy- clohexene, which is a high molecular weight impurity that normally requires a second test on an alternative column. By using an extended temperature program that employs the full thermal range of the column, 4-vinylcyclohexene, as well as all of the typical low molecular weight impurities in 1,3-butadiene, can be analyzed in a single test. Introduction 1,3-butadiene is typically isolated from products of the naphtha steam cracking process. Prior to purification, 1,3-butadiene can be contaminated with significant amounts of isobutene as well as other C4 isomers. In addition to removing these C4 isomeric contaminants during purification, it is also important that 1,3-butadiene be free of propadiene and methyl acetylene, which can interfere with catalytic polymerization. Alumina PLOT columns are the most commonly used GC column for this application, but the determination of polar hydrocarbon impurities at trace levels can be quite challenging and is highly dependent on the deactiva- tion of the alumina surface. -

Next Generation PLOT Alumina Technology for Accurate Measurement of Trace Polar Hydrocarbons in Hydrocarbon Streams
Next generation PLOT alumina technology for accurate measurement of trace polar hydrocarbons in hydrocarbon streams Jaap de Zeeuw, Tom Vezza, Bill Bromps, Rick Morehead and Gary Stidsen Restek Corporation, Bellefonte, USA 1 Next generation PLOT alumina technology for accurate measurement of trace polar hydrocarbons in hydrocarbon streams Jaap de Zeeuw, Tom Vezza, Bill Bromps, Rick More head and Gary Stidsen Restek Corporation Summary In light hydrocarbon analysis, the separation and detection of traces of polar hydrocarbons like acetylene, propadiene and methyl acetylene is very important. Using commercial alumina columns with KCl or Na2SO4 deactivation, often results in low response of polar hydrocarbons. Additionally, challenges are observed in response-in time stability. Solutions have been proposed to maximize response for components like methyl acetylene and propadiene, using alumina columns that were specially deactivated. Operating such columns showed still several challenges: Due to different deactivations, the retention and loadability of such alumina columns has been drastically reduced. A new Alumina deactivation technology was developed that combined the high response for polar hydrocarbon with maintaining the loadability. This allows the high response for components like methyl acetylene, acetylene and propadiene, also to be used for impurity analysis as well as TCD type applications. Such columns also showed excellent stability of response in time, which was superior then existing solutions. Additionally, it was observed that such alumina columns could be used up to 250 C, extending the Tmax by 50C. This allows higher hydrocarbon elution, faster stabilization and also widens application scope of any GC where multiple columns are used. In this poster the data is presented showing the improvements made in this important application field. -

BENZENE Disclaimer
United States Office of Air Quality EPA-454/R-98-011 Environmental Protection Planning And Standards June 1998 Agency Research Triangle Park, NC 27711 AIR EPA LOCATING AND ESTIMATING AIR EMISSIONS FROM SOURCES OF BENZENE Disclaimer This report has been reviewed by the Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency, and has been approved for publication. Mention of trade names and commercial products does not constitute endorsement or recommendation of use. EPA-454/R-98-011 ii TABLE OF CONTENTS Section Page LIST OF TABLES.....................................................x LIST OF FIGURES.................................................. xvi EXECUTIVE SUMMARY.............................................xx 1.0 PURPOSE OF DOCUMENT .......................................... 1-1 2.0 OVERVIEW OF DOCUMENT CONTENTS.............................. 2-1 3.0 BACKGROUND INFORMATION ...................................... 3-1 3.1 NATURE OF POLLUTANT..................................... 3-1 3.2 OVERVIEW OF PRODUCTION AND USE ......................... 3-4 3.3 OVERVIEW OF EMISSIONS.................................... 3-8 4.0 EMISSIONS FROM BENZENE PRODUCTION ........................... 4-1 4.1 CATALYTIC REFORMING/SEPARATION PROCESS................ 4-7 4.1.1 Process Description for Catalytic Reforming/Separation........... 4-7 4.1.2 Benzene Emissions from Catalytic Reforming/Separation .......... 4-9 4.2 TOLUENE DEALKYLATION AND TOLUENE DISPROPORTIONATION PROCESS ............................ 4-11 4.2.1 Toluene Dealkylation -

Supplement of Compilation and Evaluation of Gas Phase Diffusion Coefficients of Reactive Trace Gases in the Atmosphere
Supplement of Atmos. Chem. Phys., 15, 5585–5598, 2015 http://www.atmos-chem-phys.net/15/5585/2015/ doi:10.5194/acp-15-5585-2015-supplement © Author(s) 2015. CC Attribution 3.0 License. Supplement of Compilation and evaluation of gas phase diffusion coefficients of reactive trace gases in the atmosphere: Volume 2. Diffusivities of organic compounds, pressure-normalised mean free paths, and average Knudsen numbers for gas uptake calculations M. J. Tang et al. Correspondence to: M. J. Tang ([email protected]) and M. Kalberer ([email protected]) The copyright of individual parts of the supplement might differ from the CC-BY 3.0 licence. Table of Contents 1 Alkanes and cycloalkanes ............................................................................................ 1 1.1 CH 4 (methane), C 2H6 (ethane), and C 3H8 (propane) ............................................. 1 1.2 C 4H10 (butane, methyl propane) ............................................................................ 3 1.3 C 5H12 (n-pentane, methyl butane, dimethyl butane) ............................................. 5 1.4 C 6H14 (n-hexane, 2,3-dimethyl butane) ................................................................ 7 1.5 C 7H16 (n-heptane, 2,4-dimethyl pentane).............................................................. 9 1.6 C 8H18 (n-octane, 2,2,4-trimethyl pentane) .......................................................... 11 1.7 C 9H20 (n-nonane), C 10 H22 (n-decane, 2,3,3-trimethyl heptane) and C 12 H26 (n- dodecane) ................................................................................................................. -

ETHYLENE PLANT ENHANCEMENT (April 2001)
Abstract Process Economics Program Report 29G ETHYLENE PLANT ENHANCEMENT (April 2001) The prospects for ethylene demand remain strong but much of the future increase in production capacity may come from incremental enhancement of existing plants, rather than from new “grass-roots” production facilities. Steam cracking for ethylene originated as early as the 1920s and was commercialized in the 1950s. The importance of ethylene continues to drive research and development of this technology along with the exploration of nonconventional technologies in order to achieve higher yields of olefins and lower capital and operating costs. However, as recently discussed in PEP Report 29F, Ethylene by Nonconventional Processes (August 1998), it is unlikely nonconventional processes will replace steam cracking for ethylene production in the foreseeable future. This study examines potential improvements in conventional steam cracker operations with emphasis on improving the competitive edge of existing plants. Ethane, LPG and naphtha are the dominant steam cracker feedstocks. Natural gas condensate is abundant in North America and the Middle East while naphtha is commonly used in Asia and Europe. Since the 1970s many new ethylene plants have been built with feedstock flexibility. In PEP Report 220, Ethylene Feedstock Outlook (May 1999), SRIC currently projects that generally ethylene feedstocks supply will be adequate over the next decade even considering that the demand for ethylene is increasing twice as fast as petroleum refining (4- 5%/year versus 2%/year). Significant technological developments exist for all sections of the ethylene steam cracker that could be implemented in plant revamps. Examples include new large capacity yet compact and efficient furnaces; coke inhibition technology; larger, more efficient compressors; fractionation schemes; mixed refrigerants; and advance control and optimization systems. -
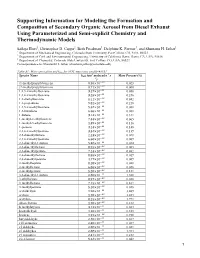
Supporting Information for Modeling the Formation and Composition Of
Supporting Information for Modeling the Formation and Composition of Secondary Organic Aerosol from Diesel Exhaust Using Parameterized and Semi-explicit Chemistry and Thermodynamic Models Sailaja Eluri1, Christopher D. Cappa2, Beth Friedman3, Delphine K. Farmer3, and Shantanu H. Jathar1 1 Department of Mechanical Engineering, Colorado State University, Fort Collins, CO, USA, 80523 2 Department of Civil and Environmental Engineering, University of California Davis, Davis, CA, USA, 95616 3 Department of Chemistry, Colorado State University, Fort Collins, CO, USA, 80523 Correspondence to: Shantanu H. Jathar ([email protected]) Table S1: Mass speciation and kOH for VOC emissions profile #3161 3 -1 - Species Name kOH (cm molecules s Mass Percent (%) 1) (1-methylpropyl) benzene 8.50×10'() 0.023 (2-methylpropyl) benzene 8.71×10'() 0.060 1,2,3-trimethylbenzene 3.27×10'(( 0.056 1,2,4-trimethylbenzene 3.25×10'(( 0.246 1,2-diethylbenzene 8.11×10'() 0.042 1,2-propadiene 9.82×10'() 0.218 1,3,5-trimethylbenzene 5.67×10'(( 0.088 1,3-butadiene 6.66×10'(( 0.088 1-butene 3.14×10'(( 0.311 1-methyl-2-ethylbenzene 7.44×10'() 0.065 1-methyl-3-ethylbenzene 1.39×10'(( 0.116 1-pentene 3.14×10'(( 0.148 2,2,4-trimethylpentane 3.34×10'() 0.139 2,2-dimethylbutane 2.23×10'() 0.028 2,3,4-trimethylpentane 6.60×10'() 0.009 2,3-dimethyl-1-butene 5.38×10'(( 0.014 2,3-dimethylhexane 8.55×10'() 0.005 2,3-dimethylpentane 7.14×10'() 0.032 2,4-dimethylhexane 8.55×10'() 0.019 2,4-dimethylpentane 4.77×10'() 0.009 2-methylheptane 8.28×10'() 0.028 2-methylhexane 6.86×10'() -

Identification of Gas-Phase Pyrolysis Products in A
Atmos. Meas. Tech., 12, 763–776, 2019 https://doi.org/10.5194/amt-12-763-2019 © Author(s) 2019. This work is distributed under the Creative Commons Attribution 4.0 License. Identification of gas-phase pyrolysis products in a prescribed fire: first detections using infrared spectroscopy for naphthalene, methyl nitrite, allene, acrolein and acetaldehyde Nicole K. Scharko1, Ashley M. Oeck1, Russell G. Tonkyn1, Stephen P. Baker2, Emily N. Lincoln2, Joey Chong3, Bonni M. Corcoran3, Gloria M. Burke3, David R. Weise3, Tanya L. Myers1, Catherine A. Banach1, David W. T. Griffith4, and Timothy J. Johnson1 1Pacific Northwest National Laboratories, Richland, WA, USA 2Rocky Mountain Research Station, USDA Forest Service, Missoula, MT, USA 3Pacific Southwest Research Station, USDA Forest Service, Riverside, CA, USA 4Centre for Atmospheric Chemistry, University of Wollongong, Wollongong NSW 2522, Australia Correspondence: Timothy J. Johnson ([email protected]) Received: 1 October 2018 – Discussion started: 13 November 2018 Revised: 16 January 2019 – Accepted: 16 January 2019 – Published: 1 February 2019 Abstract. Volatile organic compounds (VOCs) are emitted ventive tool to reduce hazardous fuel buildups in an effort to from many sources, including wildland fire. VOCs have re- reduce or eliminate the risk of such wildfires (Fernandes and ceived heightened emphasis due to such gases’ influential Botelho, 2003). Understanding the products associated with role in the atmosphere, as well as possible health effects. the burning of biomass has received considerable attention We have used extractive infrared (IR) spectroscopy on re- since the emissions can markedly impact the atmosphere. cent prescribed burns in longleaf pine stands and herein re- Fourier-transform infrared (FTIR) spectroscopy is one tech- port the first detection of five compounds using this tech- nique that has been extensively used to identify and quantify nique. -

Steam Cracking: Chemical Engineering
Steam Cracking: Kinetics and Feed Characterisation João Pedro Vilhena de Freitas Moreira Thesis to obtain the Master of Science Degree in Chemical Engineering Supervisors: Professor Doctor Henrique Aníbal Santos de Matos Doctor Štepánˇ Špatenka Examination Committee Chairperson: Professor Doctor Carlos Manuel Faria de Barros Henriques Supervisor: Professor Doctor Henrique Aníbal Santos de Matos Member of the Committee: Specialist Engineer André Alexandre Bravo Ferreira Vilelas November 2015 ii The roots of education are bitter, but the fruit is sweet. – Aristotle All I am I owe to my mother. – George Washington iii iv Acknowledgments To begin with, my deepest thanks to Professor Carla Pinheiro, Professor Henrique Matos and Pro- fessor Costas Pantelides for allowing me to take this internship at Process Systems Enterprise Ltd., London, a seven-month truly worthy experience for both my professional and personal life which I will certainly never forget. I would also like to thank my PSE and IST supervisors, who help me to go through this final journey as a Chemical Engineering student. To Stˇ epˇ an´ and Sreekumar from PSE, thank you so much for your patience, for helping and encouraging me to always keep a positive attitude, even when harder problems arose. To Prof. Henrique who always showed availability to answer my questions and to meet in person whenever possible. Gostaria tambem´ de agradecer aos meus colegas de casa e de curso Andre,´ Frederico, Joana e Miguel, com quem partilhei casa. Foi uma experienciaˆ inesquec´ıvel que atravessamos´ juntos e cer- tamente que a vossa presenc¸a diaria´ apos´ cada dia de trabalho ajudou imenso a aliviar as saudades de casa. -

Hydrocarbons Thermal Cracking Selectivity Depending on Their Structure and Cracking Parameters
Master in Chemical Engineering Hydrocarbons Thermal Cracking Selectivity Depending on Their Structure and Cracking Parameters Thesis of the Master’s Degree Development Project in Foreign Environment Cláudia Sofia Martins Angeira Erasmus coordinator: Eng. Miguel Madeira Examiner in FEUP: Eng. Fernando Martins Supervisor in ICT: Doc. Ing Petr Zámostný July 2008 Acknowledgements I would like to thank Doc. Ing. Petr Zámostný for the opportunity to hold a master's thesis in this project, the orientation, the support given during the laboratory work and suggestions for improvement through the work. i Hydrocarbons Thermal Cracking Selectivity Depending on Their Structure and Cracking Parameters Abstract This research deals with the study of hydrocarbon thermal cracking with the aim of producing ethylene, one of the most important raw materials in Chemical Industry. The main objective was the study of cracking reactions of hydrocarbons by means of measuring the selectivity of hydrocarbons primary cracking and evaluating the relationship between the structure and the behavior. This project constitutes one part of a bigger project involving the study of more than 30 hydrocarbons with broad structure variability. The work made in this particular project was focused on the study of the double bond position effect in linear unsaturated hydrocarbons. Laboratory experiments were carried out in the Laboratory of Gas and Pyrolysis Chromatography at the Department of Organic Technology, Institute of Chemical Technology, Prague, using for all experiments the same apparatus, Pyrolysis Gas Chromatograph, to increase the reliability and feasibility of results obtained. Linear octenes with different double bond position in hydrocarbon chain were used as model compounds. In order to achieve these goals, the primary cracking reactions were studied by the method of primary selectivities. -

Pahs) and SOOT FORMATION Fikret Inal A; Selim M
This article was downloaded by: [CDL Journals Account] On: 7 October 2009 Access details: Access Details: [subscription number 912375050] Publisher Taylor & Francis Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK Combustion Science and Technology Publication details, including instructions for authors and subscription information: http://www.informaworld.com/smpp/title~content=t713456315 EFFECTS OF OXYGENATE ADDITIVES ON POLYCYCLIC AROMATIC HYDROCARBONS(PAHs) AND SOOT FORMATION Fikret Inal a; Selim M. Senkan b a Department of Chemical Engineering, Izmir Institute of Technology, Urla-Izmir, Turkey. b Department of Chemical Engineering, University of California Los Angeles, Los Angeles, California, USA. Online Publication Date: 01 September 2002 To cite this Article Inal, Fikret and Senkan, Selim M.(2002)'EFFECTS OF OXYGENATE ADDITIVES ON POLYCYCLIC AROMATIC HYDROCARBONS(PAHs) AND SOOT FORMATION',Combustion Science and Technology,174:9,1 — 19 To link to this Article: DOI: 10.1080/00102200290021353 URL: http://dx.doi.org/10.1080/00102200290021353 PLEASE SCROLL DOWN FOR ARTICLE Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf This article may be used for research, teaching and private study purposes. Any substantial or systematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply or distribution in any form to anyone is expressly forbidden. The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. The accuracy of any instructions, formulae and drug doses should be independently verified with primary sources. -

SAFETY DATA SHEET Flammable Liquefied Gas Mixture: Ethane / Ethylene / Isobutane / Isobutylene / Methane / Propadiene / Propane / Propylene Section 1
SAFETY DATA SHEET Flammable Liquefied Gas Mixture: Ethane / Ethylene / Isobutane / Isobutylene / Methane / Propadiene / Propane / Propylene Section 1. Identification GHS product identifier : Flammable Liquefied Gas Mixture: Ethane / Ethylene / Isobutane / Isobutylene / Methane / Propadiene / Propane / Propylene Other means of : Not available. identification Product type : Liquefied gas Product use : Synthetic/Analytical chemistry. SDS # : 021907 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE GASES - Category 1 substance or mixture GASES UNDER PRESSURE - Liquefied gas GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable gas. May form explosive mixtures with air. Contains gas under pressure; may explode if heated. May cause frostbite. May displace oxygen and cause rapid suffocation. Precautionary statements General : Read and follow all Safety Data Sheets (SDS’S) before use. Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Close valve after each use and when empty. Use equipment rated for cylinder pressure. Do not open valve until connected to equipment prepared for use. Use a back flow preventative device in the piping. Use only equipment of compatible materials of construction. Always keep container in upright position. Approach suspected leak area with caution. Prevention : Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. No smoking. -

Safety Data Sheet
SAFETY DATA SHEET Flammable Gas Mixture: 1,3-Butadiene / Carbonyl Sulfide / Dimethyl Ether / Ethane / Hydrogen Sulfide / Isobutane / Methanol / Methyl Acetylene / Methyl Mercaptan / N- Butane / Propadiene / Propane / Propylene Section 1. Identification GHS product identifier : Flammable Gas Mixture: 1,3-Butadiene / Carbonyl Sulfide / Dimethyl Ether / Ethane / Hydrogen Sulfide / Isobutane / Methanol / Methyl Acetylene / Methyl Mercaptan / N- Butane / Propadiene / Propane / Propylene Other means of : Not available. identification Product type : Gas. Product use : Synthetic/Analytical chemistry. SDS # : 021580 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE GASES - Category 1 substance or mixture GASES UNDER PRESSURE - Compressed gas GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable gas. Contains gas under pressure; may explode if heated. May displace oxygen and cause rapid suffocation. May form explosive mixtures with air. Precautionary statements General : Read and follow all Safety Data Sheets (SDS’S) before use. Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Close valve after each use and when empty. Use equipment rated for cylinder pressure. Do not open valve until connected to equipment prepared for use. Use a back flow preventative device in the piping. Use only equipment of compatible materials of construction. Do not depend on odor to detect presence of gas.